Ministru kabineta noteikumi
Nr.600
Rīgā 2006.gada 18.jūlijā (prot.
Nr.38 44.§)
Veterināro zāļu reģistrēšanas
kārtība
Izdoti saskaņā
ar Farmācijas likuma 5.panta 3. un 6.punktu
I.Vispārīgie
jautājumi
1.Noteikumi nosaka kārtību, kādā
Zāļu valsts aģentūra reģistrē veterinārās zāles.
2.Noteikumos lietoti šādi
termini:
2.1.atsauces zāles– veterinārās
zāles, kas reģistrētas kādā Eiropas Ekonomiskās zonas valstī vai
centralizētā reģistrācijas procedūrā atbilstoši Eiropas
Parlamenta un Padomes 2004.gada 31.marta Regulai (EK)
Nr.726/2004, ar ko nosaka cilvēkiem paredzēto un veterināro zāļu
reģistrēšanas un uzraudzības Kopienas procedūras un izveido
Eiropas Zāļu aģentūru;
2.2.ģenēriskās zāles– zāles, kam
ir tāds pats aktīvo vielu kvalitatīvais un kvantitatīvais sastāvs
un tāda pati zāļu forma kā atsauces zālēm un kuru bioekvivalence
ar atsauces zālēm ir pierādīta attiecīgos biopieejamības
pētījumos. Aktīvās vielas sāļus, esterus, ēterus, izomērus,
izomēru maisījumus, kompleksos savienojumus un atvasinājumus
uzskata par to pašu aktīvo vielu, ja vien tiem nepiemīt būtiski
atšķirīgas īpašības attiecībā uz drošību un efektivitāti vai
drošību vai efektivitāti. Dažādās zāļu formas ar ātru aktīvās
vielas atbrīvošanu iekšķīgai lietošanai uzskata par vienu zāļu
formu;
2.3.gatavā veterināro zāļu sērija–
zāļu formas vienības, kas izgatavotas no viena un tā paša
sākotnējā materiāla daudzuma un kurām veikta viena un tā pati
ražošana un sterilizācija vai ražošana vai sterilizācija. Ja
ražošanas process ir nepārtraukts, gatavā veterināro zāļu sērija
ir visas noteiktā laikposmā saražotās vienības;
2.4.stiprums– aktīvās vielas
saturs kvantitatīvā izteiksmē devā, kas izteikts tilpuma vai
svara vienībā atbilstoši devas veidam;
2.5.imunoloģiskās veterinārās
zāles– veterinārās zāles, ko lieto dzīvniekiem, lai tiem
ierosinātu aktīvo vai pasīvo imunitāti vai lai diagnosticētu
imunitātes līmeni;
2.6.neimunoloģiskās veterinārās
zāles– veterinārās zāles, kas nav imunoloģiskās veterinārās
zāles;
2.7.homeopātiskās veterinārās
zāles– veterinārās zāles, kas pagatavotas no homeopātiskajām
izejvielām (produktiem, vielām vai savienojumiem) atbilstoši
homeopātisko zāļu ražošanas metodei, kura aprakstīta Eiropas
farmakopejā vai farmakopejās, ko oficiāli lieto Eiropas
Savienības dalībvalstīs (turpmāk– dalībvalstis). Homeopātisko
veterināro zāļu sastāvā var būt vairākas vielas;
2.8.zāļu izdalīšanās periods –
laikposms no pēdējās reizes, kad veterinārās zāles ievada
dzīvniekam noteiktos lietošanas apstākļos, līdz pārtikas produkta
ieguvei no šāda dzīvnieka. Šo laikposmu ievēro, lai aizsargātu
sabiedrības veselību, nodrošinot, ka minēto pārtikas produktu
sastāvā nav atliekvielu tādā daudzumā, kas pārsniedz atliekvielu
maksimāli pieļaujamo līmeni, kas noteikts Padomes 1990.gada
26.jūnija Regulā (EEK) Nr.2377/90, ar ko nosaka Kopienas
procedūru veterināro zāļu maksimāli pieļaujamo atlieku daudzuma
noteikšanai dzīvnieku izcelsmes produktos (turpmāk – Regula
Nr.2377/90);
2.9.vispārīgais nosaukums–
starptautiskais nepatentētais nosaukums, ko ieteikusi Pasaules
veselības organizācija (INN), vai, ja tāda nav, plaši lietotais
nosaukums.
3.Atļauts izplatīt veterinārās
zāles, kam Zāļu valsts aģentūra izsniegusi šo noteikumu
1.pielikumam atbilstošu veterināro zāļu reģistrācijas apliecību
(turpmāk– reģistrācijas apliecība) vai kuras reģistrētas Eiropas
Zāļu aģentūrā, veicot centralizēto reģistrācijas procedūru
atbilstoši Eiropas Parlamenta un Padomes 2004.gada 31.marta
Regulai (EK) Nr.726/2004, ar ko nosaka cilvēkiem paredzēto un
veterināro zāļu reģistrēšanas un uzraudzības Kopienas procedūras
un izveido Eiropas Zāļu aģentūru.
4.Veterinārajām zālēm, kam Zāļu
valsts aģentūra izsniegusi reģistrācijas apliecību atbilstoši šo
noteikumu prasībām, attiecībā uz papildu informāciju par šo zāļu
stiprumu, zāļu formu, ievadīšanas veidu, noformējumu un
reģistrācijas apliecības paplašinājumu (reģistrācijas apliecības
papildu attiecināšanu) Zāļu valsts aģentūra pieņem lēmumu par
zāļu pārreģistrāciju vai par datu iekļaušanu sākotnējās
reģistrācijas apliecībā. Šo lēmumu iekļauj sākotnējā lēmumā par
veterināro zāļu reģistrāciju (īpaši – attiecībā uz šo noteikumu
22., 23.un 24.punktā minētajām veterinārajām zālēm).
5.Šie noteikumi neattiecas uz:
5.1.dzīvnieku ārstniecisko barību,
kas definēta normatīvajos aktos par dzīvnieku ārstnieciskās un
diētiskās barības aprites kārtību. Jāņem vērā, ka, gatavojot
dzīvnieku ārstniecisko barību, izmanto premiksus (zāles, ko
pagatavo iepriekš, lai izgatavotu dzīvnieku ārstniecisko barību),
kas reģistrēti atbilstoši šo noteikumu prasībām;
5.2.inaktivētām imunoloģiskajām
veterinārajām zālēm, kas pagatavotas no patogēniem un antigēniem,
kuri iegūti no ganāmpulka dzīvnieka vai dzīvniekiem, un ko
izmanto minētā ganāmpulka dzīvnieka vai dzīvnieku ārstēšanai
ganāmpulkā uz vietas;
5.3.veterinārajām zālēm uz
radioaktīvo izotopu bāzes;
5.4.piedevām, kas saskaņā ar
Eiropas Parlamenta un Padomes 2003.gada 22.septembra Regulu (EK)
Nr.1831/2003 par dzīvnieku ēdināšanā lietotām piedevām ir
publicētas Eiropas Komisijas mājas lapā atļauto barības piedevu
reģistrā, ja šīs piedevas ir pievienotas dzīvnieku barībai un
dzīvnieku papildbarībai;
5.5.veterinārajām zālēm, kas
paredzētas pētījumiem un testiem veterināro zāļu izstrādes
gaitā;
5.6.veterinārajām zālēm, ko
izgatavo aptiekā atsevišķam dzīvniekam vai dzīvnieku grupai
atbilstoši veterinārārsta izrakstītai receptei, kas zināma kā
formula magistralis;
5.7.zālēm, ko izgatavo aptiekā
atbilstoši farmakopejas monogrāfijām un ko paredzēts nodot tieši
galapatērētājam, un kas zināmas kā formula
officinalis.
6.Par veterināro zāļu virzību
tirgū ir atbildīgs reģistrācijas apliecības turētājs (īpašnieks).
Pārstāvja iecelšana neatbrīvo reģistrācijas apliecības turētāju
(īpašnieku) no atbildības.
7.Veterinārās zāles, ko paredzēts
lietot produktīvajiem dzīvniekiem (dzīvnieki, no kuriem iegūst
dzīvnieku izcelsmes pārtikas produktus), reģistrē, ja to sastāvā
ietilpstošās aktīvās vielas ir norādītas Regulas Nr.2377/90 I, II
vai IIIpielikumā.
8.Reģistrācijas apliecības
turētājs (īpašnieks) vai, ja nepieciešams, Zāļu valsts aģentūra
veic nepieciešamos pasākumus, lai izdarītu grozījumus
reģistrācijas apliecībā vai anulētu reģistrācijas apliecību
atbilstoši prasībām, kas noteiktas Regulas Nr.2377/90 I, II un
IIIpielikumā. Grozījumus izdara vai reģistrācijas apliecību anulē
60dienu laikā no dienas, kad Eiropas Savienības Oficiālajā
Vēstnesī publicēti grozījumi Regulas Nr.2377/90 I, II vai III
pielikumā.
II.Kārtība, kādā
iesniedzams iesniegums veterināro zāļu reģistrācijas apliecības
saņemšanai
9.Lai saņemtu reģistrācijas
apliecību, persona, uz kā vārda paredzēts reģistrēt veterinārās
zāles (turpmāk– iesnieguma iesniedzējs), iesniedz Zāļu valsts
aģentūrā iesniegumu par veterināro zāļu reģistrāciju (turpmāk–
reģistrācijas iesniegums).
10.Reģistrācijas iesnieguma
paraugs ir publicēts Zāļu valsts aģentūras mājas lapā.
Reģistrācijas iesniegums noformēts, ievērojot Farmācijas likuma
28.1pantā minētos Eiropas Komisijas ieteikumus, kurus
Eiropas Komisija ir publicējusi Eiropas Kopienas zāļu tiesiskā
regulējuma VI sējumā “Veterinārās zāles”.
11.Reģistrācijas iesniegumu tādu
veterināro zāļu reģistrācijas apliecības saņemšanai, kas
paredzētas vienas vai vairāku sugu produktīvajiem dzīvniekiem un
kas satur aktīvās vielas, kuras attiecībā uz šīm dzīvnieku sugām
vēl nav iekļautas Regulas Nr.2377/90 I, II vai III pielikumā,
iesniedz ne agrāk kā sešus mēnešus pēc tam, kad iesniegts
atbilstoši prasībām noformēts iesniegums šīs vielas maksimāli
pieļaujamo atliekvielu daudzuma noteikšanai saskaņā ar Regulu
Nr.2377/90.
12.Reģistrācijas apliecību Zāļu
valsts aģentūra izsniedz iesnieguma iesniedzējam, kura juridiskā
adrese ir Eiropas Ekonomikas zonas valstī (reģistrēta firma,
centrālā administrācija vai faktiskā darbības vieta).
13.Iesnieguma iesniedzējs šo
noteikumu 10.punktā minētajam reģistrācijas iesniegumam pievieno
dokumentus, kuros iekļauta administratīvā informācija, un
zinātnisko dokumentāciju, kas nepieciešama, lai pierādītu
reģistrējamo veterināro zāļu drošumu, efektivitāti un kvalitāti
atbilstoši šo noteikumu 2.pielikumā (attiecībā uz
neimunoloģiskajām veterinārajām zālēm) vai 3.pielikumā (attiecībā
uz imunoloģiskajām veterinārajām zālēm) minētajām prasībām,
norādot vai pievienojot šādu informāciju:
13.1.iesnieguma iesniedzēja vārds,
uzvārds vai komersanta firmas nosaukums un faktiskā vai juridiskā
adrese. Ja atšķiras adreses, iesnieguma iesniedzējs norāda
informāciju par veterināro zāļu ražotāju vai ražotājiem, kuri
iesaistīti veterināro zāļu ražošanā, kā arī informāciju par
ražošanas telpu izvietojumu un to faktisko adresi;
13.2.veterināro zāļu nosaukums
(piešķirtais nosaukums, kas nav sajaucams ar vispārīgo
nosaukumu, vai vispārīgais vai zinātniskais nosaukums kopā ar
preču zīmi vai reģistrācijas apliecības turētāja (īpašnieka)
nosaukumu);
13.3.veterināro zāļu sastāvā
ietilpstošo aktīvo vielu un sastāvdaļu kvalitatīvie un
kvantitatīvie rādītāji. Lieto starptautisko nepatentēto
nosaukumu, ko ieteikusi Pasaules Veselības organizācija (INN), ja
tāds ir, vai aktīvo vielu ķīmisko nosaukumu;
13.4.veterināro zāļu ražošanas
metodes apraksts;
13.5.veterināro zāļu terapeitiskās
indikācijas, kontrindikācijas un blakusparādības (blaknes);
13.6.devas dzīvnieku sugām, kurām
paredzētas zāles, zāļu forma, lietošanas un ievadīšanas veids un
derīguma termiņš;
13.7.veicamie piesardzības un
drošības pasākumi, kas jāievēro, zāles uzglabājot, lietojot
zāles dzīvniekiem vai iznīcinot zāļu atkritumus. Norāda
iespējamos riskus (riska faktori, kas saistīti ar nevēlamu
ietekmi), ko zāles var radīt apkārtējai videi un cilvēku,
dzīvnieku vai augu veselībai;
13.8.periods, kādā zāles izdalās
no dzīvnieka organisma, ja zāles paredzēts lietot produktīvajiem
dzīvniekiem;
13.9.ražotāja izmantoto kontroles
metožu apraksti;
13.10.rezultāti, kas iegūti:
13.10.1.farmaceitiskajos (fizikāli
ķīmiskajos, bioloģiskajos vai mikrobioloģiskajos) testos;
13.10.2.drošuma testos un
atliekvielu testos;
13.10.3.neklīniskajos un
klīniskajos pētījumos;
13.10.4.izvērtējot veterināro zāļu
iespējamo risku apkārtējai videi. Katru iespējamā riska gadījumu
izskata atsevišķi, piedāvājot ieteikumus šī riska
mazināšanai;
13.11.sīki izstrādāts veterināro
zāļu lietošanas blakusparādību uzraudzības sistēmas apraksts un,
ja nepieciešams, sīki izstrādāts riska kontroles sistēmas
apraksts, ko iesnieguma iesniedzējs paredzējis ieviest;
13.12.veterināro zāļu apraksts
saskaņā ar šo noteikumu 35.punktu;
13.13.veterināro zāļu lietošanas
instrukcijas (turpmāk– lietošanas instrukcija) paraugs, kas
sagatavots atbilstoši normatīvajiem aktiem par veterināro zāļu
marķēšanas, izplatīšanas un kontroles noteikumiem;
13.14.primārā iepakojuma (trauks
vai citāds iepakojums, kas ir tiešā saskarē ar attiecīgajām
zālēm) un sekundārā iepakojuma (iepakojums, kurā ievieto primāro
iepakojumu) paraugi, kas sagatavoti atbilstoši normatīvajiem
aktiem par veterināro zāļu marķēšanas, izplatīšanas un kontroles
noteikumiem;
13.15.dokuments, kas apliecina, ka
veterināro zāļu ražotājam savā valstī ir atļauts ražot
veterinārās zāles;
13.16.reģistrācijas apliecību
kopijas, kas apliecina šo zāļu reģistrāciju citā Eiropas
Ekonomikas zonas valstī vai valstī, kas nav Eiropas Ekonomikas
zonas valsts (turpmāk– trešā valsts), kā arī:
13.16.1.to Eiropas Ekonomikas
zonas valstu saraksts, kurās iesniegums reģistrācijas apliecības
saņemšanai atbilstoši šo noteikumu prasībām atrodas izskatīšanas
stadijā. Šim sarakstam pievieno katrai sarakstā minētajai valstij
iesniegtās veterināro zāļu apraksta (kas sagatavots atbilstoši šo
noteikumu 35.punktam vai ko atzinusi kompetentā iestāde
atbilstoši šo noteikumu 51., 52., 53., 54.un 55.punktam) kopijas
un lietošanas instrukcijas kopijas;
13.16.2.informācija par noraidošu
lēmumu attiecībā uz veterināro zāļu reģistrācijas apliecības
izsniegšanu Eiropas Ekonomikas zonas valstī vai trešajā valstī un
šī lēmuma motivāciju;
13.16.3.šo noteikumu 13.16.1. un
13.16.2.apakšpunktā minētā informācija, kuru iesnieguma
iesniedzējs papildina, tiklīdz ir papildu ziņas par atbilstošiem
lēmumiem;
13.17.apliecinājums, ka iesnieguma
iesniedzēja rīcībā ir kvalificēta persona, kas ir atbildīga par
zāļu lietošanas izraisīto blakusparādību uzraudzības sistēmu
atbilstoši normatīvajiem aktiem par veterināro zāļu
blakusparādību uzraudzības kārtību, un ir pieejami līdzekļi, lai
paziņotu par blakusparādībām, kuras iespējams novērot Eiropas
Ekonomikas zonas valstīs vai trešajās valstīs;
13.18.attiecībā uz veterinārajām
zālēm, kas paredzētas vienas vai vairāku sugu produktīvajiem
dzīvniekiem un satur vienu vai vairākas farmakoloģiski aktīvas
vielas, kas attiecībā uz šīm dzīvnieku sugām vēl nav iekļautas
Regulas Nr.2377/90 (ar turpmākiem grozījumiem) I, II vai III
pielikumā, – dokuments, kas apliecina, ka Eiropas Zāļu aģentūrai
saskaņā ar minēto regulu ir iesniegts iesniegums maksimāli
pieļaujamo zāļu atliekvielu daudzuma noteikšanai.
14.Reģistrācijas dokumentācijā
norādītās veterināro zāļu sastāvā ietilpstošās krāsvielas
atbilst šo noteikumu 4.pielikumā minētajām prasībām.
15.Iesnieguma iesniedzējs
nodrošina, ka par šo noteikumu 13.10.apakšpunktā minēto testu un
pētījumu rezultātiem tiek sagatavots sīki izstrādāts
kopsavilkums. Kopsavilkumu sagatavo un paraksta persona
(eksperts), kuras atbilstošā tehniskā vai profesionālā
kvalifikācija izklāstīta īsā dzīves aprakstā (Curriculum
vitae). Iesnieguma iesniedzējs eksperta apstiprināto
kopsavilkumu un eksperta Curriculum vitae iesniedz Zāļu
valsts aģentūrā.
16.Ja iesnieguma iesniedzējs
atbilstoši šo noteikumu 28.punktam ir tiesīgs neiesniegt drošuma
testu un neklīnisko un klīnisko pētījumu rezultātus, eksperts
pamato zinātniskās informācijas izmantošanu saskaņā ar šo
noteikumu 2. un 3.pielikumā minētajiem nosacījumiem.
17.Kopsavilkums ir daļa no
dokumentācijas, kuru zāļu reģistrācijas iesnieguma iesniedzējs
kopā ar reģistrācijas iesniegumu iesniedz Zāļu valsts
aģentūrā.
18.Iesnieguma iesniedzējs
neiesniedz drošuma un atliekvielu testu vai neklīnisko un
klīnisko pētījumu rezultātus, ja viņš pierāda, ka reģistrējamās
veterinārās zāles ir ģenēriskās zāles atsauces zālēm, kuras
saskaņā ar šo noteikumu 3.punktu Latvijā vai citās Eiropas
Ekonomikas zonas valstīs ir reģistrētas ne mazāk kā astoņus
gadus.
19.Šo noteikumu 18.punktā minētās
veterinārās zāles neizlaiž tirgū, kamēr nav pagājuši 10 gadi pēc
atsauces zāļu sākotnējās reģistrācijas apliecības
izsniegšanas.
20.Šo noteikumu 18.punktu piemēro
arī tad, ja iesnieguma iesniedzējs iesniedzis Zāļu valsts
aģentūrā reģistrācijas iesniegumu attiecībā uz ģenēriskajām
zālēm, bet atsauces zālēm Latvijā nav izsniegta reģistrācijas
apliecība. Šajā gadījumā iesnieguma iesniedzējs norāda Eiropas
Ekonomiskās zonas valsti, kurā atsauces zāles ir vai ir bijušas
reģistrētas.Zāļu valsts aģentūra:
20.1.pieprasa Eiropas Ekonomiskās
zonas valsts, kurā atsauces zāles ir vai ir bijušas reģistrētas,
kompetentajai iestādei mēneša laikā sniegt apliecinājumu, ka
attiecīgās atsauces zāles ir vai ir bijušas reģistrētas šai
valstī, kā arī informāciju par atsauces zāļu pilnu sastāvu un, ja
nepieciešams, citus atbilstošus dokumentus;
20.2.pēc citas Eiropas Ekonomikas
zonas valsts kompetentās iestādes, kurā iesniegts veterināro zāļu
reģistrācijas iesniegums, pieprasījuma viena mēneša laikā nosūta
apstiprinājumu par to, ka atsauces zāles ir reģistrētas vai ir
bijušas reģistrētas Latvijā, un citus pieprasītos dokumentus.
21.Veterinārajām zālēm, kas
paredzētas zivīm, bitēm vai citām dzīvnieku sugām, kuras
noteiktas ar attiecīgu Eiropas Komisijas lēmumu, šo noteikumu
19.punktā noteikto 10 gadu laikposmu var pagarināt līdz 13
gadiem.
22.Iesnieguma iesniedzējs iesniedz
Zāļu valsts aģentūrā drošuma un atliekvielu testu, neklīnisko
vai klīnisko pētījumu rezultātus attiecībā uz ģenēriskajām zālēm,
ja:
22.1.uz šīm veterinārajām zālēm
nevar attiecināt šo noteikumu 2.2.apakšpunktā minēto ģenērisko
zāļu definīciju;
22.2.veicot biopieejamības
pētījumus, nevar pierādīt bioekvivalenci atsauces zālēm;
22.3.ir aktīvās vielas
terapeitisko indikāciju, stipruma, zāļu formas vai ievadīšanas
veida atšķirības attiecībā uz atsauces zālēm.
23.Ja bioloģiskās izcelsmes
veterinārās zāles, kas ir līdzīgas bioloģiskas izcelsmes atsauces
zālēm, neatbilst ģenērisku zāļu definīcijā minētajiem
nosacījumiem (īpaši– bioloģiskas izcelsmes veterināro zāļu un
bioloģiskas izcelsmes atsauces zāļu izejvielu vai ražošanas
procesa atšķirību dēļ), iesnieguma iesniedzējs iesniedz
neklīnisko vai klīnisko pētījumu rezultātus, kas attiecas uz
minētajiem nosacījumiem. Iesniedzamo papildu datu veids un
daudzums atbilst šo noteikumu 2. un 3.pielikumā minētajiem
nosacījumiem un šo noteikumu 37.punktā minētajās Eiropas
Komisijas vadlīnijās noteiktajiem kritērijiem. Pārējos atsauces
zāļu dokumentācijā iekļautos pētījumu un testu rezultātus
iesnieguma iesniedzējs ir tiesīgs neiesniegt.
24.Veterinārajām zālēm, kas
paredzētas vienas vai vairāku sugu produktīvajiem dzīvniekiem un
satur aktīvo vielu, kura Kopienā nav reģistrēta līdz 2004.gada
30.aprīlim, šo noteikumu 23.punktā minēto 10 gadu laikposmu Zāļu
valsts aģentūra pagarina par vienu gadu katram reģistrācijas
apliecības paplašinājumam attiecībā uz citām produktīvo
dzīvnieku sugām, ja šādu paplašinājumu apstiprina piecos gados no
sākotnējās reģistrācijas apliecības piešķiršanas.
25.Veterinārajām zālēm, kas
paredzētas četrām vai vairākām produktīvo dzīvnieku sugām, šo
noteikumu 24.punktā minētais laikposms kopumā nepārsniedz 13
gadus.
26.Veterinārajām zālēm, kas
paredzētas produktīvajiem dzīvniekiem, 10gadu laikposma
pagarinājumu līdz 11, 12 vai 13 gadiem piešķir tad, ja
reģistrācijas apliecības turētājs (īpašnieks) saskaņā ar Regulu
Nr.2377/90 ir sākotnēji iesniedzis Eiropas Zālu aģentūrā
iesniegumu tāda maksimāli pieļaujamā zāļu atliekvielu daudzuma
noteikšanai, kāds paredzēts dzīvnieku sugām, kuras minētas
reģistrācijas apliecībā.
27.Pētījumi un pārbaudes, kā arī
praktiskās prasības, kuras piemēro atbilstoši šo noteikumu 18.,
19., 20., 21., 22., 23.un 24.punktam, nav uzskatāmas kā pretējas
patenttiesībām vai veterināro zāļu papildu aizsardzības
sertifikātiem.
28.Iesnieguma iesniedzējs
neiesniedz drošuma un atliekvielu pārbaudes vai neklīnisko un
klīnisko pētījumu rezultātus, ja viņš pierāda, ka veterināro zāļu
sastāvā ietilpstošās aktīvās vielas Eiropas Ekonomikas zonas
valstīs ir lietotas reģistrētu veterināro zāļu sastāvā ne mazāk
kā 10 gadus, šīm zālēm ir atzīta efektivitāte un pieļaujama
drošuma pakāpe atbilstoši šo noteikumu 2.pielikumā (attiecībā uz
neimunoloģiskajām veterinārajām zālēm) vai 3.pielikumā (attiecībā
uz imunoloģiskajām veterinārajām zālēm) minētajām prasībām.
Iesnieguma iesniedzējs norāda izmantoto zinātnisko
literatūru.
29.Kā šo noteikumu 28.punktā
minēto zinātnisko literatūru (īpaši – par veterināro zāļu drošuma
pārbaudi) iesnieguma iesniedzējs var izmantot novērtējuma
ziņojumu, ko Eiropas Zāļu aģentūra publicē pēc iesnieguma
izvērtēšanas par maksimāli pieļaujamo zāļu atliekvielu daudzuma
noteikšanu saskaņā ar Regulu Nr.2377/90.
30.Ja iesnieguma iesniedzējs
izmanto zinātnisko literatūru, lai saņemtu reģistrācijas
apliecību veterinārajām zālēm, ko paredzēts lietot produktīvajiem
dzīvniekiem, un lai paplašinātu reģistrācijas apliecību (lai šīs
zāles varētu lietot citām produktīvo dzīvnieku sugām), iesnieguma
iesniedzējs atbilstoši Regulai Nr.2377/90 iesniedz jaunus
veterināro zāļu atliekvielu pētījumu rezultātus vienlaikus ar
papildu klīnisko pētījumu rezultātiem. Trešās personas šos
pētījumus un to rezultātus neizmanto trīs gadus no dienas, kad
piešķirta reģistrācijas apliecība, ar kuru saistībā izdarīti
minētie pētījumi.
31.Ja veterinārās zāles satur
aktīvās vielas, kas izmantotas reģistrētu veterināro zāļu
sastāvā, bet līdz šim nav izmantotas vienā veterināro zāļu
sastāvā terapeitiskiem nolūkiem, attiecībā uz aktīvajām vielām
iesniedz neklīnisko un klīnisko pētījumu rezultātus un, ja
nepieciešams, drošuma un atliekvielu testus.
32.Attiecībā uz šo noteikumu
31.punktā minētajām veterinārajām zālēm neiesniedz zinātniskās
atsauces par katru aktīvo vielu atsevišķi.
33.Pēc reģistrācijas apliecības
saņemšanas reģistrācijas apliecības turētājs (īpašnieks) ir
tiesīgs izmantot farmaceitisko, drošuma un atliekvielu,
neklīnisko un klīnisko dokumentāciju, kas iekļauta reģistrēto
veterināro zāļu dokumentācijā, lai izskatītu vēlāku
reģistrācijas iesniegumu veterinārajām zālēm ar tādu pašu
kvalitatīvo un kvantitatīvo aktīvo vielu sastāvu un zāļu
formu.
34.Imunoloģisko veterināro zāļu
reģistrācijas iesnieguma iesniedzējs ārkārtas apstākļos drīkst
neiesniegt tādu lauka izmēģinājumu rezultātus, kas veikti mērķa
sugai (dzīvnieku suga, kurai zāles paredzētas), ja šādus
izmeklējumus nevar veikt precīzi formulētu pamatotu iemeslu dēļ
un atbilstoši citiem Eiropas Savienības tiesību aktiem.
35.Veterināro zāļu aprakstā
iekļauj šādu informāciju (norādītajā secībā):
35.1.veterināro zāļu nosaukums,
kam pievienota norāde par stiprumu un zāļu formu;
35.2.aktīvo vielu un palīgvielu
kvalitatīvais un kvantitatīvais sastāvs, ko svarīgi zināt, lai
zāles pareizi lietotu dzīvniekiem (izmanto plaši lietotos vai
ķīmiskos nosaukumus);
35.3.zāļu forma;
35.4.klīniskie dati:
35.4.1.mērķa sugas;
35.4.2.zāļu lietošanas
indikācijas, norādot mērķa sugas;
35.4.3.kontrindikācijas;
35.4.4.blakusparādības (biežums un
smaguma pakāpe);
35.4.5.īpaši piesardzības pasākumi
attiecībā uz katru mērķa sugu;
35.4.6.zāļu lietošanas
piesardzības pasākumi, tai skaitā īpaši piesardzības pasākumi
personai, kura ievada zāles dzīvniekam;
35.4.7.veterināro zāļu lietošana
grūsnības, laktācijas vai olu dēšanas periodā;
35.4.8.veterināro zāļu
mijiedarbība ar citām zālēm un cita veida mijiedarbība;
35.4.9.ievadāmais veterināro zāļu
daudzums (deva) un ievadīšanas veids;
35.4.10.simptomi un rīcība zāļu
pārdozēšanas gadījumā (pirmā palīdzība, pretindes);
35.4.11.zāļu izdalīšanās periods
dzīvnieku izcelsmes pārtikas produktos, tai skaitā produktos,
kuros zāļu izdalīšanās periods ir noteikts nulles apmērā;
35.5.farmakoloģiskās īpašības:
35.5.1.farmakodinamiskās
īpašības;
35.5.2.farmakokinētiskie dati;
35.6.farmaceitiskie dati:
35.6.1.palīgvielu saraksts;
35.6.2.galvenās zāļu
nesaderības;
35.6.3.zāļu derīguma termiņš un
norādījumi par zāļu derīguma termiņu pēc zāļu atšķaidīšanas vai
primārā iesaiņojuma atvēršanas;
35.6.4.īpaši norādījumi zāļu
uzglabāšanai;
35.6.5.primārā iesaiņojuma veids
un saturs;
35.6.6.īpaši norādījumi rīcībai ar
neizmantotajām zālēm vai to atkritumiem, ja tādi ir;
35.7.reģistrācijas apliecības
turētājs (īpašnieks) (vārds, uzvārds vai nosaukums un juridiskā
adrese vai reģistrētā uzņēmējdarbības vieta);
35.8.reģistrācijas apliecības
numurs vai apliecību numuri;
35.9.datums, kad sākotnēji
izsniegta vai pārreģistrēta reģistrācijas apliecība;
35.10.datums, kad precizēts zāļu
apraksta teksts.
36.Iesniedzot reģistrācijai
veterinārās zāles atbilstoši šo noteikumu 18., 19., 20., 21.,
22., 23., 24., 25., 26. un 27.punktam, nav jānorāda atsauces zāļu
apraksta daļas, kas attiecas uz indikācijām vai dozējumu, uz
kurām ģenērisko zāļu tirdzniecības laikā vēl attiecas patentu
tiesības.
37.Iesnieguma iesniedzējs sagatavo
reģistrācijas iesniegumam pievienoto dokumentāciju,
ievērojot:
37.1.šo noteikumu 10., 11., 12.,
13., 14., 15., 16., 17., 18.,19., 20., 21., 22., 23., 24., 25.,
26., 27., 28., 29., 30., 31., 32., 33., 34. un 35.punktā, kā arī
2.un 3.pielikumā minētās prasības;
37.2.Farmācijas likuma
28.1pantā minētos Eiropas Komisijas ieteikumus, ko
satur Paziņojums iesniegumu iesniedzējiem tirdzniecības atļauju
saņemšanai veterinārajām zālēm Eiropas Savienības dalībvalstīs,
kuru Eiropas Komisija publicējusi Eiropas Savienības zāļu
tiesiskā regulējuma dokumentu krājuma VIsējumā “Veterinārās
zāles”;
37.3.Eiropas Komisijas
pamatnostādnes par veterināro zāļu kvalitāti, drošumu un
iedarbīgumu, kuras Eiropas Komisija publicējusi Eiropas Kopienas
Zāļu tiesiskā reglamentējuma dokumentu krājumā;
37.4.nosacījumu, ka ir iesniegta
informācija par reģistrējamo veterināro zāļu novērtēšanu
(neatkarīgi no tā, vai novērtēšana ir zālēm labvēlīga vai
nelabvēlīga). Iesnieguma iesniedzējs iesniedz Zāļu valsts
aģentūrā arī informāciju par veterināro zāļu testiem vai
pētījumu, kas ir nepilnīgs vai nav pabeigts. Pēc reģistrācijas
apliecības saņemšanas reģistrācijas apliecības turētājs
(īpašnieks) Zāļu valsts aģentūrai bez kavēšanās sniedz jebkādu
informāciju, kas nav iekļauta sākotnējā reģistrācijas
dokumentācijā un attiecas uz ieguvuma un riska novērtējumu
(veterināro zāļu pozitīvās terapeitiskās iedarbības izvērtējums
saistībā ar noteiktiem riskiem);
37.5.nosacījumu, ka eksperimentus
ar dzīvniekiem veic atbilstoši normatīvajiem aktiem par
eksperimentos un zinātniskos nolūkos izmantojamo dzīvnieku
turēšanas, izmantošanas, tirdzniecības un nogalināšanas
kārtību.
III.Kārtība,
kādā reģistrē homeopātiskās veterinārās zāles
38.Šī nodaļa neattiecas uz
imunoloģiskajām homeopātiskajām veterinārajām zālēm.
39.Zāļu valsts aģentūra:
39.1.homeopātiskās veterinārās
zāles, kuras ražo un izplata Latvijā, reģistrē saskaņā ar šo
noteikumu prasībām;
39.2.homeopātiskajām veterinārajām
zālēm, kas atbilst šo noteikumu 40.punkta prasībām:
39.2.1.izveido vienkāršotu
reģistrācijas procedūru;
39.2.2.piemēro šo noteikumu 73.,
74., 75., 76., 77., 78., 79., 80.un 81.punktu.
40.Zāļu valsts aģentūra piemēro
vienkāršoto reģistrācijas procedūru homeopātiskajām veterinārajām
zālēm, kuru:
40.1.ievadīšanas veids atbilst
Eiropas Farmakopejas monogrāfijai vai, ja tāda nav, Eiropas
Ekonomikas zonas valstu oficiāli lietoto farmakopeju
monogrāfijām;
40.2.marķējumā un lietošanas
instrukcijā vai kādā no reģistrācijai iesniegtajiem dokumentiem
nav norādītas specifiskas terapeitiskās indikācijas;
40.3.atšķaidīšanas pakāpe ir
pietiekami augsta, lai nodrošinātu zāļu drošumu.Zāles nesatur
vairāk kā vienu 10000 daļu no mātes tinktūras
(pamattinktūras).
41.Reģistrējot homeopātiskās
veterinārās zāles, Zāļu valsts aģentūra klasificē tās recepšu
vai bezrecepšu (zāles, kuras atļauts izplatīt bez receptes)
zālēs.
42.Šo noteikumu 40.punktā
minētajām homeopātiskajām veterinārajām zālēm vienkāršotajā
reģistrācijas procedūrā izmanto kritērijus un nosacījumus
atbilstoši šo noteikumu IVnodaļā minētajām prasībām. Nepiemēro šo
noteikumu 51., 52., 53., 54. un 55.punktā minētās prasības un
zāļu terapeitisko iedarbību apliecinošo dokumentu noformēšanas
prasības.
43.Iesnieguma iesniedzējs, kurš
reģistrē homeopātiskās veterinārās zāles, piemērojot vienkāršoto
reģistrācijas procedūru, iesniedz Zāļu valsts aģentūrā
reģistrācijas iesniegumu. Vienkāršotā reģistrācijas iesniegumā
var iekļaut homeopātiskās veterinārās zāles, kas iegūtas no
vienas un tās pašas homeopātiskās izejvielas (izejvielām).
Homeopātisko veterināro zāļu reģistrācijas iesnieguma
iesniedzējs, lai apliecinātu homeopātisko veterināro zāļu
kvalitāti un saražoto sēriju homogenitāti (zāļu sēriju
identiskumu), reģistrācijas iesniegumam pievieno:
43.1.dokumentu, kurā aprakstītas
homeopātiskās izejvielas vai izejvielu zinātniskais nosaukums,
vai farmakopejā norādītais nosaukums kopā ar aprakstu par zāļu
ievadīšanas veidiem, zāļu formām un atšķaidījuma pakāpi;
43.2.aprakstu par homeopātiskās
izejvielas vai izejvielu iegūšanu, kontroli un to homeopātisko
iedarbību, izmantojot atbilstošu bibliogrāfiju. Ja homeopātiskās
veterinārās zāles satur bioloģiskas vielas, apraksta veiktos
pasākumus zāļu nodrošināšanai pret patogēniem
mikroorganismiem;
43.3.dokumentu par katras zāļu
formas ražošanu un kontroli;
43.4.atšķaidīšanas un potencēšanas
metodes aprakstu;
43.5.speciālo atļauju (licenci)
attiecīgo homeopātisko veterināro zāļu ražošanai;
43.6.citās Eiropas Ekonomikas
zonas valstīs izsniegto reģistrācijas apliecību kopijas, kas
izsniegtas tām pašām homeopātiskajām veterinārajām zālēm;
43.7.vienu vai vairākus
reģistrējamo homeopātisko veterināro zāļu primārā un sekundārā
iepakojuma paraugus vai to reklāmas paraugus;
43.8.dokumentu, kas apliecina
homeopātisko veterināro zāļu stabilitāti;
43.9.ieteikto zāļu izdalīšanās
laiku un tā noteikšanas pamatojumu.
44.Vienkāršoto reģistrācijas
procedūru nepiemēro homeopātiskajām veterinārajām zālēm, kas nav
minētas šo noteikumu 40.punktā (kas paredzētas produktīvo
dzīvnieku ārstēšanai). Reģistrācijas apliecības izsniegšanas
procedūra šīm homeopātiskajām veterinārajām zālēm ir atbilstoša
šo noteikumu 10., 11., 12., 13., 14., 15., 16., 17., 18.,19.,
20., 21., 22., 23., 24., 25., 26., 27., 28., 29., 30., 31., 32.,
33., 34., 35., 36. un 37.punktā minētajām prasībām.
45.Zāļu valsts aģentūra ir tiesīga
piemērot izņēmumus attiecībā uz drošuma pārbaudi un klīniskiem un
neklīniskiem pētījumiem homeopātiskajām veterinārajām zālēm,
kuras paredzēts lietot mājas (istabas) dzīvniekiem un
neproduktīvajiem eksotiskajiem dzīvniekiem saskaņā ar Latvijā
praktizētajiem homeopātijas principiem un raksturīgajām iezīmēm.
Šajā gadījumā Zāļu valsts aģentūra paziņo Eiropas Komisijai par
piemērotajiem izņēmumiem.
IV.Kārtība, kādā
izskatāms reģistrācijas iesniegums un izsniedzama veterināro zāļu
reģistrācijas apliecība
46.Zāļu valsts aģentūra izsniedz
veterināro zāļu reģistrācijas apliecību 210 dienu laikā no
dienas, kad saņemts atbilstoši šiem noteikumiem noformēts
reģistrācijas iesniegums. Reģistrācijas iesniegumus, kas par
vienām veterinārajām zālēm iesniegti divās vai vairākās Eiropas
Ekonomikas zonas valstīs, izskata saskaņā ar šo noteikumu V
nodaļā minētajām prasībām.
47.Zāļu valsts aģentūra ir tiesīga
pārtraukt reģistrācijas iesnieguma izskatīšanu, ja reģistrācijas
iesniegums un dokumentācija jau tiek izskatīta citā Eiropas
Ekonomikas zonas valstī. Šādā gadījumā Zāļu valsts aģentūra
informē iesnieguma iesniedzēju, ka tā piemēro šo noteikumu V
nodaļā minētās prasības.
48.Ja Zāļu valsts aģentūra saskaņā
ar šo noteikumu 13.4.1.apakšpunktu saņem informāciju, ka kādā
Eiropas Ekonomikas zonas valstī reģistrācijas apliecību ir
saņēmušas veterinārās zāles, par kurām Zāļu valsts aģentūrā
iesniegts reģistrācijas iesniegums, Zāļu valsts aģentūra noraida
reģistrācijas iesniegumu, ja vien tas nav iesniegts atbilstoši šo
noteikumu Vnodaļā minētajām prasībām.
49.Zāļu valsts aģentūra izskata
reģistrācijas iesniegumu, kas iesniegts saskaņā ar šo noteikumu
10., 11., 12., 13., 14., 15., 16., 17., 18.,19., 20., 21., 22.,
23., 24., 25., 26., 27., 28., 29., 30., 31., 32., 33. un
34.punktā minētajām prasībām, un:
49.1.pārbauda informāciju, kas
norādīta reģistrācijas iesniegumam pievienotajos dokumentos;
49.2.ir tiesīga nosūtīt gatavās
veterinārās zāles, to izejvielas un, ja nepieciešams,
starpproduktus vai citas zāļu sastāvdaļas laboratoriskai
izmeklēšanai Zāļu valsts aģentūras laboratorijā vai citā
laboratorijā, kura ir tiesīga kontrolēt zāles, lai pārbaudītu,
vai iesniegtajos reģistrācijas dokumentos zāļu ražotāja norādītās
zāļu kontroles metodes ir pietiekamas veterināro zāļu
kvalitatīvai kontrolei;
49.3.pieprasa, lai iesnieguma
iesniedzējs sniedz papildu informāciju, ja iesniegtā informācija
nav pilnīga.Šo noteikumu 46.punktā minēto reģistrācijas laiku
pagarina līdz brīdim, kad Zāļu valsts aģentūrā iesniegti
pieprasītie dokumenti vai, ja nepieciešams, iesnieguma
iesniedzējs sniedzis mutisku vai rakstisku paskaidrojumu uz
jautājumiem, kas radušies, izskatot iesniegtos reģistrācijas
dokumentus;
49.4.ir tiesīga pieprasīt, lai
iesnieguma iesniedzējs Pārtikas un veterinārajā dienestā
iesniedz nepieciešamās vielas vajadzīgajos daudzumos, lai
pārbaudītu analītiskās metodes to veterināro zāļu atliekvielu
noteikšanai, kuras iesnieguma iesniedzējs norādījis
reģistrācijas dokumentos.
50.Zāļu valsts aģentūra:
50.1.pārbauda, vai veterināro zāļu
ražotājs spēj ražot veterinārās zāles saskaņā ar ziņām, kas
iesniegtas saskaņā ar šo noteikumu 13.4.apakšpunktu, un veikt
veterināro zāļu kvalitātes kontroli ar metodēm, kas norādītas
reģistrācijai iesniegtajos dokumentos saskaņā ar šo noteikumu
13.9.apakšpunktu. Ja atsevišķos pamatotos gadījumos veterināro
zāļu ražotājam dažus veterināro zāļu ražošanas posmus vai
veterināro zāļu kvalitātes kontroli izpilda trešā persona, Zāļu
valsts aģentūra ir tiesīga veikt pārbaudi attiecīgajā
uzņēmumā;
50.2.pārbauda, vai veterināro zāļu
importētājs no trešajām valstīm spēj veikt veterināro zāļu
kvalitātes kontroli ar metodēm, kas norādītas reģistrācijai
iesniegtajos dokumentos saskaņā ar šo noteikumu 13.9.apakšpunktu.
Ja atsevišķos pamatotos gadījumos veterināro zāļu importētājam no
trešajām valstīm veterināro zāļu kvalitātes kontroli izpilda
trešā persona, Zāļu valsts aģentūra ir tiesīga veikt pārbaudi
attiecīgajā uzņēmumā;
50.3.pārbaudot trešajās valstīs
ražoto veterināro zāļu atbilstību šo noteikumu
50.1.apakšpunktam, ņem vērā kārtību, kas noteikta Eiropas
Komisijas apkopojumā par Kopienas inspicēšanas un informācijas
apmaiņas procedūrām.
51.Zāļu valsts aģentūra izsniedz
reģistrācijas apliecību un apstiprina zāļu aprakstu. Par
apstiprināto zāļu aprakstu Zāļu valsts aģentūra informē
reģistrācijas apliecības turētāju (īpašnieku).
52.Zāļu valsts aģentūra nodrošina,
ka informācija par veterinārajām zālēm (īpaši – zāļu marķējums un
lietošanas instrukcija) atbilst zāļu aprakstam, kuru Zāļu valsts
aģentūra apstiprinājusi vienlaikus ar reģistrācijas apliecību vai
pēc tam.
53.Zāļu valsts aģentūra
nekavējoties nodrošina reģistrēto veterināro zāļu reģistrācijas
apliecības un zāļu apraksta publisku pieejamību Zāļu valsts
aģentūras mājas lapā internetā.
54.Zāļu valsts aģentūra sagatavo
veterināro zāļu novērtējuma ziņojumu, kurā iekļauj komentārus par
reģistrācijai iesniegtajiem dokumentiem (tai skaitā par
veterinārajām zālēm veikto farmakoloģisko, drošuma un atliekvielu
testu un neklīnisko un klīnisko pētījumu rezultātiem). Veterināro
zāļu novērtējuma ziņojumu papildina ar jaunu informāciju par
veterināro zāļu kvalitāti, drošumu un iedarbību, tiklīdz šāda
informācija kļūst pieejama.
55.Zāļu valsts aģentūra izņem no
šo noteikumu 54.punktā minētā veterināro zāļu novērtējuma
ziņojuma komerciāli konfidenciālu informāciju un publisko pārējo
ziņojuma tekstu Zāļu valsts aģentūras mājas lapā internetā.
56.Zāļu valsts aģentūra ir tiesīga
pieprasīt, lai reģistrācijas apliecības turētājs (īpašnieks) uz
primārā vai sekundārā iepakojuma un lietošanas instrukcijā
norāda informāciju par piesardzības pasākumiem, kurus ievēro,
lietojot zāles dzīvniekam, vai citu cilvēku un dzīvnieku
veselības aizsardzībai vai drošībai nepieciešamu informāciju,
kas kļuvusi zināma pēc farmakoloģisko testu vai klīnisko pētījumu
rezultātiem vai zāļu izmantošanas veterinārmedicīniskajā
praksē.
57.Izņēmuma gadījumos objektīvu un
pārbaudāmu iemeslu dēļ Zāļu valsts aģentūra pēc konsultēšanās ar
iesnieguma iesniedzēju ir tiesīga pieņemt lēmumu par veterināro
zāļu reģistrāciju, ja iesnieguma iesniedzējs izpilda noteiktas
prasības (īpaši – attiecībā uz zāļu drošumu), kā arī paziņo par
starpgadījumiem saistībā ar veterināro zaļu lietošanu un
veiktajiem pasākumiem. Reģistrācijas apliecība paliek spēkā, ja
Zāļu valsts aģentūra katru gadu atkārtoti izvērtē prasību
izpildi.
58.Pēc reģistrācijas apliecības
saņemšanas tās turētājs (īpašnieks), ņemot vērā zinātnes un
tehnikas progresu, papildina veterināro zāļu kontroles metodes,
lai veterinārās zāles tiktu ražotas un pārbaudītas atbilstoši
vispāratzītām zinātniskām metodēm. Par izmaiņām informē Zāļu
valsts aģentūru. Zāļu valsts aģentūra izvērtē un apstiprina
izmaiņas.
59.Reģistrācijas apliecības
turētājs (īpašnieks) pēc Pārtikas un veterinārā dienesta
pieprasījuma:
59.1.nodrošina pietiekamu daudzumu
vielu, lai varētu veikt veterināro zāļu atliekvielu noteikšanu
atbilstoši noteikumiem par zāļu atliekvielu noteikšanu un tās
finansēšanas kārtību;
59.2.pamatojoties uz savu
pieredzi, sniedz tehniskās konsultācijas, lai atvieglotu
veterināro zāļu atliekvielu noteikšanas analīzes metodes
ieviešanu Pārtikas un veterinārā dienesta Nacionālajā
diagnostikas centrā.
60.Reģistrācijas apliecības
turētājs (īpašnieks) nekavējoties informē Zāļu valsts aģentūru,
ja ir pieejama informācija:
60.1.kuras dēļ jāpapildina ziņas
vai dokumenti, kas noformēti atbilstoši šo noteikumu 13., 18.,
19., 20., 21., 22., 23., 24., 25., 26., 27., 28., 29., 30., 31.,
32., 35., 36. un 37.punktā vai šo noteikumu 2. un 3.pielikumā
minētajām prasībām;
60.2.par kompetento iestāžu
noteiktajiem ierobežojumiem vai aizliegumiem attiecībā uz
veterinārajām zālēm katrā valstī, kurā šīs veterinārās zāles
laistas tirdzniecībā;
60.3.kas var ietekmēt šo
veterināro zāļu riska un ieguvuma attiecības izvērtēšanu. Lai
turpinātu riska un ieguvuma attiecības izvērtējumu, Zāļu valsts
aģentūra ir tiesīga pieprasīt, lai reģistrācijas apliecības
turētājs (īpašnieks) sniedz informāciju, kas apliecina, ka riska
un ieguvuma attiecība saglabājas labvēlīga.
61.Reģistrācijas apliecības
turētājs (īpašnieks) nekavējoties informē Zāļu valsts aģentūru,
ja nepieciešami grozījumi šo noteikumu 10., 11., 12., 13., 14.,
15., 16., 17., 18.,19., 20., 21., 22., 23., 24., 25., 26., 27.,
28., 29., 30., 31., 32., 33. un 34.punktā minētajā veterināro
zāļu reģistrācijas dokumentācijā vai reģistrācijas apliecībā.
62.Pēc reģistrācijas apliecības
saņemšanas tās turētājs (īpašnieks) informē Zāļu valsts aģentūru
par:
62.1.dienu, kad reģistrētās
veterinārās zāles laiž tirgū, ņemot vērā reģistrētās zāļu
formas;
62.2.laikposmu, uz kuru pārtraukta
veterināro zāļu laišanu tirgū vai veterināro zāļu izņemšana no
tirgus. Minēto informāciju reģistrācijas apliecības turētājs
(īpašnieks) paziņo ne vēlāk kā divus mēnešus pirms dienas, kad
tiek pārtraukta veterināro zāļu laišana tirgū, izņemot ārkārtējus
apstākļus.
63.Pēc Zāļu valsts aģentūras
pieprasījuma reģistrācijas apliecības turētājs (īpašnieks)
atbilstoši veterināro zāļu lietošanas izraisīto blakusparādību
uzraudzības sistēmai norāda visus datus par veterināro zāļu
pārdošanas apjomu un viņa rīcībā esošos datus par izrakstīto
recepšu apjomu.
64.Reģistrācijas apliecība ir
derīga piecus gadus.
65.Reģistrācijas apliecību pēc
pieciem gadiem pārreģistrē, pamatojoties uz riska un ieguvuma
attiecības atkārtotu izvērtējumu. Reģistrācijas apliecības
turētājs (īpašnieks) iesniedz Zāļu valsts aģentūrā iesniegumu un
tam pievienotu konsolidētu sarakstu, kurā norādīti dokumenti, kas
iesniegti par veterināro zāļu kvalitāti, drošumu un efektivitāti,
un visi grozījumi, kas ieviesti kopš reģistrācijas apliecības
sākotnējās izsniegšanas. Reģistrācijas apliecības turētājs
(īpašnieks) iesniegumu un konsolidēto sarakstu iesniedz Zāļu
valsts aģentūrā ne vēlāk kā sešus mēnešus pirms reģistrācijas
apliecības derīguma termiņa beigām. Zāļu valsts aģentūra katrā
laikā ir tiesīga pieprasīt, lai reģistrācijas apliecības turētājs
(īpašnieks) iesniedz sarakstā norādītos dokumentus, lai precizētu
iesniegto informāciju.
66.Reģistrācijas apliecība pēc
pārreģistrēšanas saskaņā ar šo noteikumu 65.punktu ir derīga
neierobežotu laiku (izņemot gadījumus, ja Zāļu valsts aģentūra,
pamatojoties uz veterināro zāļu lietošanas izraisīto
blakusparādību uzraudzības sistēmas rezultātiem, nosaka vienu
papildu pārreģistrāciju pēc pieciem gadiem saskaņā ar šo
noteikumu 65.punktu).
67.Reģistrācijas apliecība
uzskatāma par nederīgu, ja veterinārās zāles triju gadu laikā pēc
lēmuma par attiecīgo veterināro zāļu reģistrāciju pieņemšanas
dienas nav laistas tirgū.
68.Reģistrācijas apliecība
uzskatāma par nederīgu, ja pēc veterināro zāļu reģistrācijas tās
ir laistas tirgū, bet pēc tam trīs gadus pēc kārtas netiek
tirgotas.
69.Zāļu valsts aģentūra, ņemot
vērā ārkārtas apstākļus un cilvēku vai dzīvnieku veselības
aizsardzības apsvērumus, ir tiesīga izdarīt izņēmumus attiecībā
uz šo noteikumu 66. un 67.punktā minētajiem nosacījumiem.Šādus
izņēmumus attiecīgi pamato.
70.Reģistrācijas apliecības
izsniegšana nemazina veterināro zāļu ražotāja un reģistrācijas
apliecības turētāja (īpašnieka) atbildību.
71.Zāļu valsts aģentūra
reģistrācijas apliecību neizsniedz, ja:
71.1.izskatot veterināro zāļu
reģistrācijai iesniegtos dokumentus, tiek konstatēts, ka
reģistrācijai iesniegtie dokumenti neatbilst šo noteikumu 10.,
11., 12., 13., 14., 15., 16., 17., 18.,19., 20., 21., 22., 23.,
24., 25., 26., 27., 28., 29., 30., 31., 32., 33. un 34.punktā
minētajām prasībām;
71.2.pēc reģistrācijai iesniegto
dokumentu un datu pārbaudes tiek konstatēts, ka:
71.2.1.veterināro zāļu riska un
ieguvuma attiecība, tās lietojot reģistrācijas dokumentos
norādītajā veidā, ir nelabvēlīga (īpaši – vērtējot ieguvumus
saistībā ar dzīvnieku veselību un labturību un patērētāju drošību
attiecībā uz veterinārajām zālēm, ko lieto zootehniskos
nolūkos);
71.2.2.veterinārajām zālēm nav
reģistrācijas dokumentācijā norādītās terapeitiskās iedarbības
vai iesnieguma iesniedzējs nav sniedzis pietiekamus pierādījumus
par šādu iedarbību reģistrācijas dokumentos norādītajām dzīvnieku
mērķa sugām;
71.2.3.veterināro zāļu
kvalitatīvais un kvantitatīvais sastāvs neatbilst reģistrācijas
dokumentos norādītajiem datiem;
71.2.4.iesnieguma iesniedzēja
ieteiktais veterināro zāļu izdalīšanās periods:
71.2.4.1.nav pietiekami ilgs, lai
nodrošinātu, ka no ārstētajiem dzīvniekiem iegūtie dzīvnieku
izcelsmes pārtikas produkti nesatur veterināro zāļu atliekvielas
patērētāja veselībai bīstamos daudzumos;
71.2.4.2.nav pietiekami
pamatots;
71.2.5.iesnieguma iesniedzēja
piedāvātais veterināro zāļu marķējums vai lietošanas instrukcija
neatbilst prasībām, kas noteiktas veterināro zāļu marķēšanas,
izplatīšanas un kontroles noteikumos;
71.2.6.veterināro zāļu lietošanas
veids ir aizliegts Eiropas Savienībā.
72.Iesnieguma iesniedzējs vai
reģistrācijas atļaujas turētājs ir atbildīgs par iesniegto datu
pareizību.
V.Kārtība, kādā
veterinārās zāles reģistrē, izmantojot savstarpējās atzīšanas
procedūru un decentralizēto procedūru
73.Lai zāles reģistrētu vairāk
nekā vienā Eiropas Ekonomikas zonas valstī (turpmāk – iesaistītās
dalībvalstis), iesnieguma iesniedzējs katras iesaistītās
dalībvalsts kompetentajā iestādē (Latvijā – Zāļu valsts aģentūrā)
iesniedz veterināro zāļu reģistrācijas iesniegumu, izpildot šādus
nosacījumus:
73.1.veterināro zāļu reģistrācijas
dokumentācijā ietver administratīvo informāciju un zinātnisko un
tehnisko informāciju atbilstoši prasībām, kas minētas šo
noteikumu 10., 11., 12., 13., 18., 19., 20., 21., 22., 23., 24.,
25., 26., 27., 28., 29., 30., 31., 32., 35., 36. un
37.punktā;
73.2.vienādu reģistrācijas
dokumentāciju iesniedz visās iesaistītajās dalībvalstīs;
73.3.reģistrācijas dokumentācijai
pievieno to iesaistīto dalībvalstu sarakstu, kurās iesniegts
reģistrācijas iesniegums.
74.Ja iesaistītajās dalībvalstīs
veterinārās zāles reģistrācijas iesnieguma iesniegšanas
laikā:
74.1.ir reģistrētas, veterinārās
zāles reģistrējamas savstarpējās atzīšanas procedūrā;
74.2.atrodas reģistrācijas
procesā, veterinārās zāles reģistrējamas decentralizētā
procedūrā.
75.Pirms iesnieguma iesniegšanas
par veterināro zāļu reģistrāciju, izmantojot savstarpējās
atzīšanas procedūru, iesnieguma iesniedzējs pieprasa kādai
iesaistītajai dalībvalstij, lai tā ir atsauces dalībvalsts un
sagatavo veterināro zāļu novērtējuma ziņojumu. Ja atsauces
dalībvalsts ir Latvija, Zāļu valsts aģentūra sagatavo novērtējuma
ziņojumu par attiecīgajām veterinārajām zālēm vai atjauno
novērtējuma ziņojumu Administratīvā procesa likumā noteiktajā
kārtībā, bet ne ilgāk kā 90 dienu laikā pēc prasībām atbilstoša
reģistrācijas iesnieguma saņemšanas. Ja nepieciešams, Zāļu valsts
aģentūra novērtējuma ziņojumā iekļauj novērtējumu atbilstoši šo
noteikumu 26., 27. un 28.punktā minētajām prasībām vai 32.punktā
minētajām prasībām. Novērtējuma ziņojumu ar apstiprinātu
veterināro zāļu aprakstu, marķējumu un lietošanas instrukciju
Zāļu valsts aģentūra nosūta iesnieguma iesniedzējam un
iesaistītajām dalībvalstīm.
76.Ja līdz reģistrācijas
iesnieguma iesniegšanas dienai reģistrācijas apliecība citā
iesaistītajā dalībvalstī nav izsniegta un Latvija ir atsauces
dalībvalsts, iesnieguma iesniedzējs pieprasa, lai Zāļu valsts
aģentūra sagatavo novērtējuma ziņojuma projektu un veterināro
zāļu apraksta, marķējuma un lietošanas instrukcijas projektu.
Zāļu valsts aģentūra sagatavo minēto projektu Administratīvā
procesa likumā noteiktajā kārtībā, bet ne ilgāk kā 120 dienu
laikā pēc derīga reģistrācijas iesnieguma saņemšanas un nosūta to
iesaistītajām dalībvalstīm un iesnieguma iesniedzējam.
77.Zāļu valsts aģentūra 90 dienu
laikā pēc šo noteikumu 75. vai 76.punktā minētā novērtējuma
ziņojuma projekta un zāļu apraksta, marķējuma teksta un
lietošanas instrukcijas projekta saņemšanas:
77.1.apstiprina minētos dokumentus
un informē par to atsauces dalībvalsti, ja Latvija ir iesaistītā
dalībvalsts;
77.2.pēc informācijas saņemšanas
no iesaistītajām dalībvalstīm reģistrē iesaistīto dalībvalstu
vienošanos, noslēdz procedūru un nekavējoties informē par to
iesnieguma iesniedzēju, ja Latvija ir atsauces dalībvalsts.
78.Ja Zāļu valsts aģentūrā
iesniegts iesniegums par veterināro zāļu reģistrāciju saskaņā ar
šo noteikumu 73.punktu, Zāļu valsts aģentūra:
78.1.pieņem lēmumu atbilstoši
apstiprinātajam novērtējuma ziņojumam, zāļu aprakstam, marķējuma
tekstam un lietošanas instrukcijai Administratīvā procesa likumā
noteiktajā kārtībā, bet ne vēlāk kā 30 dienu laikā pēc šo
noteikumu 77.2.apakšpunktā minētās vienošanās
apstiprināšanas;
78.2.par pieņemto lēmumu informē
atsauces dalībvalsti, reģistrācijas iesnieguma iesniedzēju un
citas iesaistītās dalībvalstis.
79.Ja Zāļu valsts aģentūra,
pamatojoties uz to, ka reģistrējamās veterinārās zāles var
apdraudēt cilvēku vai dzīvnieku veselību vai apkārtējo vidi,
90dienu laikā neapstiprina novērtējuma ziņojumu, zāļu aprakstu,
marķējuma tekstu un lietošanas instrukciju, Zāļu valsts
aģentūra:
79.1.iesniedz sīki pamatotus
apsvērumus iesnieguma iesniedzējam, atsauces dalībvalstij un
iesaistītajām dalībvalstīm;
79.2.par jautājumiem, kuros nav
panākta vienošanās, nekavējoties informē Koordinācijas grupu,
kura izveidota Eiropas Zāļu aģentūrā, lai izskatītu visus
jautājumus, kas saistīti ar zāļu reģistrāciju divās vai vairākās
Eiropas Ekonomikas zonas valstīs (turpmāk– Koordinācijas grupa).
Koordinācijas grupas sekretariāta darbu nodrošina Eiropas Zāļu
aģentūra.
80.Lai pamatotu atteikumu izsniegt
imunoloģisko veterināro zāļu reģistrācijas apliecību
decentralizētajā procedūrā, Zāļu valsts aģentūra ir tiesīga
piemērot nosacījumus par aizliegumu izplatīt imunoloģiskās
veterinārās zāles, kas noteikti normatīvajos aktos par veterināro
zāļu marķēšanas, izplatīšanas un kontroles noteikumiem.
81.Zāļu valsts aģentūra 60dienu
laikā pēc šo noteikumu 79.2.apakšpunktā minētās informācijas
saņemšanas nodrošina iesnieguma iesniedzējam iespēju sniegt
mutisku vai rakstisku paskaidrojumu.
82.Ja iesaistītās dalībvalstis par
jautājumiem, kuros nav panākta vienošanās, 60 dienu laikā pēc
paziņošanas Koordinācijas grupai:
82.1.vienojas:
82.1.1.un Latvija ir atsauces
dalībvalsts, Zāļu valsts aģentūra reģistrē vienošanos, slēdz
procedūru un informē par to iesnieguma iesniedzēju;
82.1.2.un Latvija ir iesaistītā
dalībvalsts, Zāļu valsts aģentūra pieņem lēmumu atbilstoši šo
noteikumu 78.punktā minētajai kārtībai;
82.2.nevienojas un Latvija ir
atsauces dalībvalsts, Zāļu valsts aģentūra nekavējoties iesniedz
Eiropas Zāļu aģentūrā sīki izstrādātu faktu aprakstu par
jautājumu, par kuru netika panākta vienošanās, un domstarpību
iemeslus. Šo dokumentu kopiju nosūta iesnieguma iesniedzējam.
83.Tiklīdz iesnieguma iesniedzējs
ir informēts par nevienošanos un saņēmis šo noteikumu
82.2.apakšpunktā minēto dokumentu kopiju, iesnieguma iesniedzējs
nekavējoties nosūta Eiropas Zāļu aģentūrai šo noteikumu 73.punktā
minētās informācijas un dokumentu kopijas.
84.Ja Zāļu valsts aģentūra ir
saņēmusi divus vai vairākus iesniegumus konkrētu veterināro zāļu
reģistrācijai saskaņā ar šo noteikumu 10., 11., 12., 13., 18.,
19., 20., 21., 22., 23., 24., 25., 26., 27., 28., 29., 30., 31.,
32., 35., 36. un 37.punktā minēto kārtību un citas Eiropas
Ekonomikas zonas valstis ir pieņēmušas atšķirīgus lēmumus
attiecībā uz šo veterināro zāļu reģistrāciju, reģistrācijas
apliecības apturēšanu vai anulēšanu, Zāļu valsts aģentūra,
Eiropas Komisija vai reģistrācijas apliecības turētājs
(īpašnieks) ir tiesīgs vērsties Veterināro zāļu komitejā
(turpmāk– Komiteja), identificēt problēmu un iesniegt visu tā
rīcībā esošo informāciju.
85.Zāļu valsts aģentūra atbilstoši
Eiropas Zāļu aģentūras un Eiropas Komisijas lēmumam 22 dienu
laikā pēc strīda izskatīšanas var nosūtīt rakstisku atzinumu par
Eiropas Komisijas lēmuma projektu. Zāļu valsts aģentūra ir
tiesīga iesniegt rakstisku lūgumu, lai lēmuma projektu apspriež
Komitejā. Ja pēc Eiropas Komisijas atzinuma Zāļu valsts aģentūras
rakstiskais atzinums izraisa jaunus svarīgus zinātniskas un
tehnoloģiskas dabas jautājumus, kam nav pievērsta uzmanība,
dokumenta apspriešana un izskatīšana Eiropas Zāļu aģentūrā tiek
turpināta.
86.Zāļu valsts aģentūra un
atsauces dalībvalsts kompetentā iestāde pieņem lēmumu par
veterināro zāļu reģistrāciju vai reģistrācijas apliecības
anulēšanu, vai nepieciešamību grozīt reģistrācijas dokumentāciju,
lai izpildītu Eiropas Komisijas lēmumu 30 dienu laikā pēc Eiropas
Komisijas lēmuma paziņošanas. Zāļu valsts aģentūra par pieņemto
lēmumu informē Eiropas Komisiju un Eiropas Zāļu aģentūru.
87.Reģistrācijas apliecības
turētājs (īpašnieks), kas reģistrējis veterinārās zāles
savstarpējās atzīšanas procedūrā vai decentralizētajā procedūrā,
jebkurus grozījumus (izmaiņas) veterināro zāļu reģistrācijas
apliecībā, kas piešķirta saskaņā ar šajā nodaļā minētajām
prasībām, iesniedz visām Eiropas Ekonomikas zonas valstīm, kurās
attiecīgas veterinārās zāles reģistrētas.
88.Ja Zāļu valsts aģentūra cilvēku
un dzīvnieku veselības un dabas aizsardzības apsvērumu dēļ
uzskata, ka:
88.1.ir nepieciešami grozījumi
reģistrācijas apliecībā, kas izsniegta saskaņā ar šajā nodaļā
minētajām prasībām, vai ka reģistrācijas apliecību ir
nepieciešams apturēt vai atsaukt, tā nekavējoties vēršas Eiropas
Zāļu aģentūrā, identificē problēmu un iesniedz visu tās rīcībā
esošo attiecīgo informāciju;
88.2.ir nepieciešama steidzama
rīcība, lai aizsargātu cilvēku vai dzīvnieku veselību vai vidi,
Zāļu valsts aģentūra Latvijas teritorijā ir tiesīga apturēt
saskaņā ar šajā nodaļā minētajām prasībām izsniegto veterināro
zāļu reģistrācijas apliecības darbību. Zāļu valsts aģentūra ne
vēlāk kā nākamajā darbdienā informē par savas rīcības iemesliem
Eiropas Komisiju un citas Eiropas Ekonomiskās zonas valstis,
kurās ir reģistrētas šīs veterinārās zāles.
89.Šo noteikumu 81., 82., 83.,
84., 85. un 86.punktā minētās prasības nepiemēro šo noteikumu
40.punktā minētajām homeopātiskajām veterinārajām zālēm.
90.Šo noteikumu 73., 74., 75.,
76., 77., 78., 79., 80., 81., 82., 83., 84. un 85.punktā minētās
prasības nepiemēro šo noteikumu 45.punktā minētajām
homeopātiskajām veterinārajām zālēm.
VI.Veterināro
zāļu klīniskā izpēte un lietošanas novērojumi
91.Ja iesnieguma iesniedzējs
veterināro zāļu klīnisko izpēti un lietošanas novērojumus veic
Latvijas Republikā, to veikšanai jāsaņem Pārtikas un veterinārā
dienesta atļauja.
92.Iesnieguma iesniedzējs
nodrošina veterināro zāļu klīniskās izpētes un lietošanas
novērojumu veikšanu atbilstoši šo noteikumu 37.punktā, kā arī 2.
un 3.pielikumā minētajām prasībām.
93.Veterināro zāļu klīniskajā
izpētē un lietošanas novērojumos iegūtos datus iesnieguma
iesniedzējs nosūta Zāļu valsts aģentūrai. Dati ir pietiekami, lai
sniegtu zinātniski pamatotu atzinumu par veterināro zāļu
atbilstību veterināro zāļu reģistrācijas kritērijiem.
VII.Izmaiņas
veterināro zāļu reģistrācijas dokumentācijā un reģistrācijas
paplašināšana
94.Lai apstiprinātu izmaiņas
veterināro zāļu reģistrācijas dokumentācijā, reģistrācijas
apliecības turētājs (īpašnieks) iesniedz iesniegumu par izmaiņu
apstiprināšanu. Iesniegumam pievieno datus un dokumentus, kas
atbilst Eiropas Komisijas publicētajam paraugam un ir sagatavoti
saskaņā ar šajos noteikumos noteiktajām prasībām reģistrācijas
dokumentācijai. Minēto iesniegumu un dokumentus iesniedz:
94.1.Zāļu valsts aģentūrā par
veterinārajām zālēm, kas reģistrētas nacionālā reģistrācijas
procedūrā un ko nereģistrē savstarpējā atzīšanas procedūrā vai
decentralizētā procedūrā;
94.2.Zāļu valsts aģentūrā un
attiecīgo Eiropas Ekonomikas zonas valstu kompetentajās iestādēs
saskaņā ar Eiropas Komisijas 2003.gada 3.jūnija Regulu (EK)
Nr.1084/2003 par izmaiņu izskatīšanu tās tirdzniecības atļaujas
nosacījumos, ko dalībvalsts kompetenta iestāde izsniegusi
attiecībā uz cilvēkiem paredzētām zālēm un veterinārām zālēm, ja
zāles ir reģistrētas savstarpējā atzīšanas procedūrā vai
decentralizētā procedūrā;
94.3.Eiropas Zāļu aģentūrā saskaņā
ar Komisijas 2003.gada 3.jūnija Komisijas Regulu (EK)
Nr.1085/2003 par izmaiņu izskatīšanu cilvēkiem paredzētu zāļu un
veterināro zāļu tirdzniecības atļaujas nosacījumos, uz ko
attiecas Padomes Regula (EEK) Nr.2309/93, ja zāles ir reģistrētas
centralizētā reģistrācijas procedūrā.
95.Zāļu valsts aģentūra atbilstoši
kompetencei izpilda šo noteikumu 94.3.apakšpunktā minētajā
regulā noteiktos kompetentās uzraudzības iestādes pienākumus.
96.Lai saņemtu lēmumu par
veterināro zāļu reģistrācijas paplašināšanu nacionālā
reģistrācijas procedūrā reģistrētām zālēm (ko nereģistrē
savstarpējā atzīšanas procedūrā vai decentralizētā procedūrā),
reģistrācijas apliecības turētājs (īpašnieks) iesniedz Zāļu
valsts aģentūrā iesniegumu saskaņā ar Eiropas Komisijas publicēto
paraugu, kas ir publicēta Zāļu valsts aģentūras mājas lapā
internetā (kurā ir iekļauti dati un ir norādīti pievienotie
dokumenti).
97.Attiecībā uz nacionālā
reģistrācijas procedūrā reģistrētām veterinārajām zālēm (ko
nereģistrē savstarpējā atzīšanas procedūrā vai decentralizētā
procedūrā) Zāļu valsts aģentūra:
97.1.pieņem lēmumu par veterināro
zāļu reģistrācijas paplašināšanu, ja iesniegtie dati un dokumenti
apliecina atbilstību šo noteikumu 5.pielikumā minētajiem
kritērijiem. Šādā gadījumā veterināro zāļu nosaukums atbilst
veterināro zāļu nosaukumam, kas norādīts sākotnējā lēmumā par
zāļu reģistrāciju;
97.2.saskaņā ar šo noteikumu VIII
vai IX nodaļu izvērtē iesniegto iesniegumu par izmaiņu
apstiprināšanu un pieņem lēmumu par izmaiņu apstiprināšanu vai
neapstiprināšanu.
VIII.Nelielas IA
un IB tipa izmaiņas veterināro zāļu reģistrācijas
dokumentācijā
98.Nacionālā reģistrācijas
procedūrā reģistrētām veterinārajām zālēm (ko nereģistrē
savstarpējā atzīšanas procedūrā vai decentralizētā procedūrā)
nelielas I tipa (IA tipa un IB tipa) izmaiņas reģistrācijas
dokumentācijā ir minētas šo noteikumu 6.pielikumā. Minētās
izmaiņas novērtē un apstiprina saskaņā ar šo nodaļu.
99.Šo noteikumu 98.punktā
minētajām veterinārajām zālēm:
99.1.iesniegumā par nelielām I
tipa izmaiņām norāda tikai vienas I tipa izmaiņas;
99.2.vienu iesniegumu, norādot
tajā saistību starp visām izrietošajām izmaiņām, iesniedz,
ja:
99.2.1.no vienām IA tipa izmaiņām
izriet viena vai vairākas citas IA tipa izmaiņas;
99.2.2.no vienām IB tipa izmaiņām
izriet IA tipa vai IB tipa izmaiņas;
99.3.ja izdarāmas vairākas I tipa
izmaiņas, par katru izmaiņu iesniedz atsevišķu iesniegumu. Katrā
iesniegumā ir atsauce uz pārējiem iesniegumiem, ja tādi ir;
99.4.ja no I tipa izmaiņām izriet,
ka zāļu aprakstā, marķējumā vai lietošanas instrukcijā jāizdara
grozījumi, to uzskata par izmaiņu daļu.
100.Zāļu valsts aģentūra izskata
iesnieguma atbilstību šo noteikumu 98.un 99.punktā minētajiem
nosacījumiem, novērtē iesniegtos datus un dokumentus (dokumentu
ekspertīze) un Administratīvā procesa likumā noteiktajā kārtībā
pieņem lēmumu par izmaiņu apstiprināšanu reģistrācijas
dokumentācijā vai izmaiņu neapstiprināšanu, ievērojot, ka datu un
dokumentu novērtēšana (ekspertīze) tiek veikta:
100.1.nelielām IA tipa izmaiņām –
14dienu laikā pēc iesnieguma saņemšanas Zāļu valsts
aģentūrā;
100.2.nelielām IB tipa izmaiņām –
30dienu laikā pēc iesnieguma saņemšanas Zāļu valsts
aģentūrā.
101.Zāļu valsts aģentūra ir
tiesīga pieprasīt, lai reģistrācijas apliecības turētājs
(īpašnieks) sniedz papildu informāciju. Šādā gadījumā šo
noteikumu 100.punktā minēto procedūru aptur līdz dienai, kad
reģistrācijas apliecības turētājs (īpašnieks) iesniedz Zāļu
valsts aģentūrā pieprasīto papildu informāciju.
102.Ja pēc iesnieguma par IB tipa
izmaiņām un tam pievienoto datu, dokumentu un citas informācijas
novērtēšanas Zāļu valsts aģentūra uzskata, ka minētais iesniegums
nav pieņemams, Zāļu valsts aģentūra Administratīvā procesa likumā
noteiktajā kārtībā, bet ne ilgāk kā 30 dienu laikā pēc šo
noteikumu 98.punktā minētā iesnieguma saņemšanas sniedz
reģistrācijas apliecības turētājam (īpašniekam) pamatotu
atzinumu. Reģistrācijas apliecības turētājs (īpašnieks) ir
tiesīgs 30 dienu laikā pēc Zāļu valsts aģentūras atzinuma
saņemšanas izdarīt grozījumus iesniegumā par IB tipa izmaiņām,
ievērojot Zāļu valsts aģentūras atzinumā minēto pamatojumu. Ja
reģistrācijas apliecības turētājs (īpašnieks) izdara minētos
grozījumus iesniegumā par IB tipa izmaiņām, tas minēto 30dienu
laikā rakstiski paziņo Zāļu valsts aģentūrai par grozījumu
veikšanu.
103.Ja reģistrācijas apliecības
turētājs (īpašnieks) iesniegumā par IB tipa izmaiņām neizdara
grozījumus saskaņā ar šo noteikumu 73.punktā minētajiem
nosacījumiem, iesniegumu uzskata par noraidītu, un Zāļu valsts
aģentūra pieņemto lēmumu par izmaiņu neapstiprināšanu paziņo
reģistrācijas apliecības turētājam (īpašniekam) Administratīvā
procesa likumā noteiktajā kārtībā, bet ne ilgāk kā 30 dienu
laikā.
104.Ja Zāļu valsts aģentūra nav
paziņojusi reģistrācijas apliecības turētājam (īpašniekam)
lēmumu par izmaiņu neapstiprināšanu atbilstoši šo noteikumu
102.punktā minētajai kārtībai, ir uzskatāms, ka Zāļu valsts
aģentūra izmaiņas ir apstiprinājusi un reģistrācijas apliecības
turētājs (īpašnieks) var ieviest paredzētās izmaiņas.
IX.Nozīmīgas II
tipa izmaiņas reģistrācijas dokumentācijā un reģistrācijas
apliecības turētāja (īpašnieka) firmas vai adreses maiņa
105.Nacionālā reģistrācijas
procedūrā reģistrētām veterinārajām zālēm (ko nereģistrē
savstarpējā atzīšanas procedūrā vai decentralizētā procedūrā)
nozīmīgas - II tipa izmaiņas – ir izmaiņas, kas:
105.1.neatbilst šo noteikumu 5. un
6.pielikumam;
105.2.ir saistītas ar
reģistrācijas apliecības turētāja (īpašnieka) firmas maiņu
(esošais reģistrācijas apliecības turētājs (īpašnieks) un jaunais
reģistrācijas apliecības turētājs (īpašnieks) nav viena un tā
pati persona) vai adreses maiņu.
106.Šo noteikumu 105.punktā
minētajām veterinārajām zālēm:
106.1.iesniegumā par II tipa
izmaiņām norāda tikai vienas II tipa izmaiņas, izņemot gadījumu,
ja:
106.1.1.par vienām zālēm izdara
vairākas II tipa izmaiņas.Šādā gadījumā par katru izmaiņu
iesniedz atsevišķu iesniegumu, un katrā iesniegumā ir atsauce uz
pārējiem iesniegumiem, ja tādi ir;
106.1.2.no vienām II tipa izmaiņām
izriet citas izmaiņas. Par visām šādām izmaiņām var iesniegt
vienu iesniegumu, un tajā norāda saistību starp visām
izrietošajām izmaiņām;
106.2.ja no izmaiņām izriet, ka
veterināro zāļu aprakstā, marķējumā vai lietošanas instrukcijā
jāizdara grozījumi, to uzskata par izmaiņu daļu.
107.Zāļu valsts aģentūra izskata
iesnieguma atbilstību šo noteikumu 105.un 106.punktā minētajiem
nosacījumiem, novērtē iesniegtos datus un dokumentus (dokumentu
ekspertīze) un Administratīvā procesa likumā noteiktajā kārtībā
pieņem lēmumu par izmaiņu apstiprināšanu reģistrācijas
dokumentācijā. Datus un dokumentus novērtē (ekspertīzi veic):
107.1.ne ilgāk kā 60 dienu laikā
pēc iesnieguma saņemšanas Zāļu valsts aģentūrā. Minēto termiņu
var samazināt, ja jautājums ir steidzams (īpaši – saistībā ar
drošības jautājumiem);
107.2.ne ilgāk kā 90 dienu laikā
pēc iesnieguma saņemšanas Zāļu valsts aģentūrā, ja iesniegums
saistīts ar terapeitisko indikāciju izmaiņām vai indikāciju
papildināšanu.
108.Zāļu valsts aģentūra ir
tiesīga pieprasīt, lai reģistrācijas apliecības turētājs
(īpašnieks) sniedz papildu informāciju. Šādā gadījumā šo
noteikumu 107.punktā minēto procedūru aptur līdz dienai, kad
reģistrācijas apliecības turētājs (īpašnieks) iesniedz Zāļu
valsts aģentūrā pieprasīto papildu informāciju.
109.Nacionālā reģistrācijas
procedūrā reģistrētām veterinārajām zālēm (ko nereģistrē
savstarpējā atzīšanas procedūrā vai decentralizētā procedūrā)
reģistrācijas apliecības turētāja (īpašnieka) nosaukuma vai
juridiskās adreses maiņas gadījumā esošais reģistrācijas
apliecības turētājs (īpašnieks) iesniedz Zāļu valsts aģentūrā
iesniegumu, ja reģistrācijas apliecības turētājs (īpašnieks) nav
tā pati persona.Iesniegumam pievieno šādus dokumentus un norāda
šādu informāciju:
109.1.dokumentu, kas apliecina
reģistrācijas apliecības turētāja (īpašnieka) maiņu;
109.2.veterināro zāļu nosaukumu,
reģistrācijas apliecības numuru un reģistrācijas datumu;
109.3.esošā reģistrācijas
apliecības turētāja (īpašnieka) firmas nosaukumu un adresi, kā
arī tās personas nosaukumu un adresi, kurai paredzēts nodot zāļu
reģistrācijas apliecību;
109.4.dokumentu, kas apliecina
pilnīgas un nodošanas brīdī atjaunotas veterināro zāļu
reģistrācijas dokumentācijas vai tās kopijas pieejamību un
nodošanu personai, kurai nodod zāļu reģistrācijas apliecību;
109.5.datumu, kad persona, kurai
paredzēts nodot reģistrācijas apliecību, pārņem pienākumus no
iepriekšējā reģistrācijas apliecības turētāja (īpašnieka);
109.6.dokumentu, kas apliecina, ka
persona, kurai nodod reģistrācijas apliecību, spēj veikt
reģistrācijas apliecības turētāja (īpašnieka) pienākumus
atbilstoši prasībām, kas noteiktas normatīvajos aktos par
veterināro zāļu ražošanu un kontroli, veterināro zāļu
izplatīšanu, reklāmu, nevēlamām blakusparādībām un klīnisko
izpēti. Minētajā dokumentā norāda:
109.6.1.personu, kura ir atbildīga
par darbībām saistībā ar veterināro zāļu blakusparādību
pārraudzības sistēmu (personas adrese, tālruņa un faksa numurs,
īss darbības un pieredzes apraksts);
109.6.2.dienestu, kurš ir
atbildīgs par veterināro zāļu reklamēšanu un izplatīšanu
(dienesta adrese, tālruņa un faksa numurs);
109.7.veterināro zāļu apraksta,
lietošanas instrukcijas un marķējuma projektu;
109.8.dokumentu, kas apliecina
maksājumu par novērtēšanu saskaņā ar Zāļu valsts aģentūras
publisko pakalpojumu cenrādi.
110.Datumu, kad reģistrācijas
apliecība tiks nodota jaunajam reģistrācijas apliecības turētājam
(īpašniekam), nosaka Zāļu valsts aģentūra, un šo datumu ieraksta
līgumā, kuru slēdz esošais un jaunais reģistrācijas apliecības
turētājs (īpašnieks). Reģistrācijas apliecības darbības termiņš
nemainās.
X.Noslēguma
jautājumi
111.Atzīt par spēku zaudējušiem
Ministru kabineta 2004.gada 22.aprīļa noteikumus Nr.409
“Veterināro zāļu reģistrācijas kārtība” (Latvijas Vēstnesis,
2004, 69., 201.nr.; 2006, 68.nr.).
112.Veterināro zāļu reģistrācijas
apliecības, kas izsniegtas līdz šo noteikumu spēkā stāšanās
dienai, ir derīgas līdz tajās norādītā derīguma termiņa
beigām.
Informatīva
atsauce uz Eiropas Savienības direktīvām
Noteikumos iekļautas tiesību
normas, kas izriet no:
1)Eiropas Parlamenta un Padomes
2001.gada 6.novembra Direktīvas 2001/82/EK par Kopienas kodeksu,
kas attiecas uz veterinārajām zālēm;
2)Eiropas Parlamenta un Padomes
2004.gada 31.marta Direktīvas 2004/28/EK, kas labo Direktīvu
2001/82/EK par Kopienas kodeksu, kas attiecas uz veterinārajām
zālēm.
Ministru
prezidents A.Kalvītis
Zemkopības
ministra vietā – iekšlietu ministrs Dz.Jaundžeikars
Redakcijas
piebilde: noteikumi stājas spēkā ar 2006.gada 11.augustu.
Zemkopības
ministrijas iesniegtajā redakcijā
1.pielikums
Ministru kabineta
2006.gada 18.jūlija noteikumiem
Nr.600

Zemkopības ministra vietā –
iekšlietu ministrs Dz.Jaundžeikars
Zemkopības ministrijas iesniegtajā redakcijā
2.pielikums
Ministru kabineta
2006.gada 18.jūlija noteikumiem
Nr.600
Prasības
neimunoloģiskajām veterinārajām zālēm
I.
Dokumentācijas kopsavilkums
1. Administratīvie dati:
1.1. veterinārās zāles, uz kurām
attiecas reģistrācijas iesniegums, identificē ar nosaukumu un
aktīvās vielas (aktīvo vielu) nosaukumu, norādot zāļu stiprumu,
zāļu formu, ievadīšanas veidu un lietošanas veidu, kā arī galīgās
realizācijas noformējuma aprakstu;
1.2. uzrāda reģistrācijas
iesnieguma iesniedzēja nosaukumu un adresi, ražotāju nosaukumus,
adreses un objektus, kas iesaistīti dažādos ražošanas posmos,
ietverot galaprodukta ražotāju un aktīvās vielas ražotāju, un
attiecīgā gadījumā importētāja nosaukumu un adresi;
1.3. reģistrācijas iesnieguma
iesniedzējs precīzi norāda to dokumentu skaitu un nosaukumu,
kurus viņš iesniedz kopā ar iesniegumu, kā arī norāda uz
iesniegtajiem paraugiem, ja tādus iesniedz;
1.4. administratīvajiem datiem
pielikumā pievieno dokumentu, kurā atspoguļots, ka ražotājam ir
atļauts ražot attiecīgās veterinārās zāles, kā noteikts
normatīvajos aktos par kārtību, kādā izsniedz, aptur, pārreģistrē
un anulē speciālās atļaujas (licences) veterinārfarmaceitiskai
darbībai (tirdzniecības atļauja), kā arī to Eiropas Ekonomikas
zonas valstu (turpmāk– dalībvalstu) sarakstu, kurās ir piešķirta
reģistrācijas apliecība, kopijas no visiem zāļu aprakstiem
saskaņā ar šo noteikumu 35.punkta prasībām, ko apstiprinājušas
dalībvalstis, un to dalībvalstu sarakstu, kurās iesniegts
pieteikums.
2. Zāļu apraksts:
2.1. iesnieguma iesniedzējs
piedāvā zāļu aprakstu saskaņā ar šo noteikumu 35.punkta
prasībām;
2.2. iesnieguma iesniedzējs uzrāda
vienu vai vairākus veterināro zāļu paraugus vai reklāmas maketus
kopā ar lietošanas instrukcijas paraugu, ja tā nepieciešama.
3. Eksperta kopsavilkuma
ziņojums:
3.1. saskaņā ar šo noteikumu 15.,
16. un 17.punkta prasībām iesniedz eksperta kopsavilkuma ziņojumu
par analīzes dokumentāciju, farmaceitiski toksikoloģisko
dokumentāciju, atliekvielu dokumentāciju un klīnisko
dokumentāciju;
3.2. katru kopsavilkuma ziņojumu
veido dažādo testu un/vai pētījumu, kas veikti saskaņā ar šo
noteikumu prasībām, kritisks izvērtējums, un tajā uzrāda visus
datus, kas attiecas uz izvērtējumu. Eksperts sniedz savu atzinumu
par to, vai ir pietiekamas garantijas attiecīgo veterināro zāļu
kvalitātei, drošumam un iedarbīgumam. Faktu kopsavilkums vien ir
nepietiekams;
3.3. visus svarīgos datus apkopo
eksperta kopsavilkuma ziņojuma pielikumā, ja iespējams, tabulas
vai grafiskā veidā. Eksperta ziņojumā un kopsavilkumos ir
precīzas savstarpējās norādes uz ziņām, ko satur
pamatdokumenti;
3.4. katru eksperta kopsavilkuma
ziņojumu sagatavo persona ar atbilstošu kvalifikāciju un
pieredzi. To paraksta un datē eksperts, un kopsavilkuma ziņojumam
pievieno īsas ziņas par eksperta izglītību un profesionālo
pieredzi. Atspoguļo eksperta un reģistrācijas iesnieguma
iesniedzēja profesionālās attiecības.
II.
Neimunoloģisko veterināro zāļu analītiskās (fizikāli ķīmiskās,
bioloģiskās vai mikrobioloģiskās) pārbaudes
4. Visas testēšanas procedūras
atbilst tā brīža zinātnes progresa stāvoklim un ir atzītas
procedūras, kā arī tiek sniegti apstiprināšanas pētījumu
rezultāti.
5. Visas testēšanas procedūras
apraksta pietiekami sīki, lai to varētu atkārtot kontroles
pārbaudēs, ko veic pēc Zāļu valsts aģentūras pieprasījuma. Sīki
apraksta izmantojamās speciālās iekārtas un aprīkojumu, pēc
iespējas pievienojot shēmu. Vajadzības gadījumā laboratorijas
reaģentu formulas papildina ar pagatavošanas metodi. Tādām
testēšanas procedūrām, kas iekļautas Eiropas Farmakopejā vai
dalībvalsts farmakopejā, šo aprakstu var aizstāt ar sīki
izstrādātu atsauci uz minēto farmakopeju.
6. Šo noteikumu 13.3.apakšpunktā
noteiktās ziņas par veterināro zāļu sastāvā ietilpstošo aktīvo
vielu un sastāvdaļu kvalitatīvo un kvantitatīvo sastāvu iesniedz
saskaņā ar šādām prasībām:
6.1. kvalitatīvās ziņas (zāļu visu
komponentu kvalitatīvo ziņu jēdziens ietver sevī to, ka tiek
nosauktas un aprakstītas aktīvās vielas, palīgvielu komponenti
neatkarīgi no to veida un izmantotā daudzuma, ietverot
krāsvielas, konservantus, palīgvielas, stabilizatorus,
biezinātājus, emulgatorus, garšas un aromātiskās vielas u.tml.,
zāļu ārējā apvalka (kapsulu, želatīna kapsulu u.tml.) komponenti,
ko paredzēts caur gremošanas traktu vai kā citādi ievadīt
dzīvniekiem). Šīm ziņām pievieno visus datus attiecībā uz trauku
un to noslēgšanas veidu kopā ar datiem par zāļu lietošanas un
ievadīšanas ierīcēm, ko piegādā kopā ar šīm zālēm;
6.2. “parastā terminoloģija”, kas
lietojama, aprakstot zāļu komponentus, neatkarīgi no šo noteikumu
13.3.apakšpunkta pārējo noteikumu piemērošanas, nozīmē:
6.2.1. attiecībā uz vielām, kas
ierakstītas Eiropas Farmakopejā vai, ja tās nav, kādas
dalībvalsts farmakopejā– galveno nosaukumu attiecīgās
monogrāfijas augšdaļā ar atsauci uz attiecīgo farmakopeju;
6.2.2. attiecībā uz pārējām
vielām– Pasaules Veselības organizācijas ieteikto starptautisko
nepatentēto nosaukumu (SNN), kam var būt pievienots kāds cits
nepatentēts nosaukums, vai, ja tāda nav, precīzu zinātnisko
nosaukumu. Vielas, kam nav starptautiska nepatentēta nosaukuma
vai precīza zinātniskā nosaukuma, apraksta, nosakot, kā un no kā
tās pagatavotas, attiecīgi papildinot ar citām atbilstošām
ziņām;
6.2.3. attiecībā uz krāsvielām–
“E” koda apzīmējumu, kas tam piešķirts atbilstoši šo noteikumu
4.pielikuma prasībām;
6.3. kvantitatīvās ziņas:
6.3.1. lai sniegtu zāļu visu
aktīvo vielu “kvantitatīvās ziņas”, atbilstoši zāļu formai nosaka
katras aktīvās vielas masu vai bioloģiskās aktivitātes vienību
skaitu devas vienībā vai masas, vai tilpuma vienībā;
6.3.2. bioloģiskās aktivitātes
vienību izmanto vielām, ko nevar noteikt ķīmiski. Ja Pasaules
Veselības organizācija ir noteikusi Starptautisku bioloģiskās
aktivitātes vienību, tad lieto to. Ja nav noteikta Starptautiska
bioloģiskās aktivitātes vienība, tad bioloģiskās aktivitātes
vienības izsaka tādā veidā, lai sniegtu nepārprotamu informāciju
par vielu aktivitāti;
6.3.3. ja vien iespējams, norāda
bioloģisko aktivitāti masas vai tilpuma vienībā. Šai informācijai
pievieno:
6.3.3.1. attiecībā uz injicējamām
veterinārajām zālēm– katras trauka vienībā esošās aktīvās vielas
masu vai bioloģiskās aktivitātes vienības, ņemot vērā zāļu
izmantojamo apjomu pēc atjaunošanas;
6.3.3.2. attiecībā uz tādām zālēm,
ko ievada pilienos– katras aktīvās vielas masu vai bioloģiskās
aktivitātes vienības, kas ir tādā pilienu skaitā, kurš atbilst 1
ml vai 1 g pagatavojuma;
6.3.3.3. attiecībā uz sīrupiem,
emulsijām, granulām un citām farmaceitiskajām formām, ko ievada
nomērītos daudzumos– katras aktīvās vielas masu vai bioloģiskās
aktivitātes vienības nomērītajā daudzumā;
6.3.4. aktīvās vielas, kas ir
savienojumu vai atvasinājumu veidā, apraksta kvantitatīvi
attiecībā uz to kopējo masu un, ja nepieciešams vai ir
lietderīgi, attiecībā uz molekulas aktīvā elementa vai elementu
masu;
6.3.5. zālēm, kurās ir aktīvā
viela, uz kuru pirmo reizi attiecas reģistrācijas iesniegums
jebkurā dalībvalstī, tādas aktīvās vielas kvantitatīvo
novērtējumu, kas ir sālis vai hidrāts, sistemātiski izsaka ar
aktīvā elementa vai elementu masu molekulā. Visām pēc tam
reģistrētajām zālēm dalībvalstīs to kvantitatīvo sastāvu deklarē
tādā pašā veidā attiecībā uz to pašu aktīvo vielu.
7. Farmaceitiskā izstrādne:
7.1. sniedz skaidrojumu par
savienojumu, komponentu un trauka izvēli un paredzēto palīgvielas
funkciju gatavajās zālēs;
7.2. šim skaidrojumam pievieno
zinātniskos datus par farmaceitisko izstrādni. Nosaka devas
palielinājumu, to pamatojot.
8. Ražošanas metodes apraksts:
8.1. šo noteikumu 13.4.apakšpunktā
minēto veterināro zāļu ražošanas metodes aprakstu izveido tādu,
lai tas sniegtu pietiekamu pārskatu par ražošanas operāciju
raksturu;
8.2. aprakstā iekļauj:
8.2.1. norādi uz dažādajiem
ražošanas posmiem, lai būtu iespējams veikt novērtējumu attiecībā
uz to, vai farmaceitiskās formas ražošanā lietotais process
varētu izraisīt nevēlamas pārmaiņas komponentos;
8.2.2. nepārtrauktās ražošanas
gadījumā– pilnīgas ziņas par piesardzības pasākumiem, kas veikti,
lai nodrošinātu galaprodukta viendabību;
8.2.3. faktisko ražošanas formulu,
sniedzot sīki izklāstītas kvantitatīvās ziņas par visām
izmantotajām vielām. Papildvielu daudzumus uzrāda aptuveni,
ciktāl tas vajadzīgs attiecībā uz farmaceitisko formu. Uzskaita
visas vielas, kas varētu zust ražošanas gaitā. Norāda jebkādu
devas palielinājumu un to pamato;
8.2.4. norādi par ražošanas
posmiem, kuros veic paraugu ņemšanu kontrolei ražošanas gaitā, ja
pārējie dati reģistrācijas iesniegumam pievienotajos dokumentos
rāda šādas kontroles nepieciešamību gatavo zāļu kvalitātes
kontrolei;
8.2.5. eksperimentālu pētījumu
rezultātus, kas apstiprina ražošanas procesu, ja izmanto
nestandarta ražošanas metodi vai ja tam ir izšķirīga nozīme
attiecībā uz zālēm;
8.2.6. attiecībā uz sterilām
zālēm– ziņas par lietotajiem sterilizācijas procesiem un/vai
dezinfekcijas pasākumiem.
9. Izejmateriālu kontrole:
9.1. šajā punktā “izejmateriāli”
nozīmē jebkādu medicīnisko produktu sastāvu un, vajadzības
gadījumā, tā trauka komponentus, kā minēts šī pielikuma
6.1.apakšpunktā;
9.2. gadījumā, ja tā ir:
9.2.1. aktīva viela, kas nav
aprakstīta Eiropas Farmakopejā vai dalībvalsts farmakopejā;
9.2.2. aktīva viela, kas ir
aprakstīta Eiropas Farmakopejā vai dalībvalsts farmakopejā, bet
kuras pagatavošanas metode var atstāt piemaisījumus, kas nav
minēti farmakopejas monogrāfijā un kuru atbilstīgai kvalitātes
kontrolei monogrāfija nav lietderīga. Ja to ražojusi cita
persona, nevis reģistrācijas iesnieguma iesniedzējs, tad
reģistrācijas iesnieguma iesniedzējs var organizēt to, lai
aktīvās vielas ražotājs nepastarpināti iesniegtu kompetentajām
iestādēm sīki izstrādātu aprakstu attiecībā uz ražošanas metodi,
kvalitātes kontroli ražošanas gaitā un procesa apstiprināšanu.
Šajā gadījumā ražotājs sniedz reģistrācijas iesnieguma
iesniedzējam visas ziņas, kas nepieciešamas, lai reģistrācijas
iesnieguma iesniedzējs uzņemtos atbildību par šīm zālēm. Ražotājs
rakstiski apstiprina reģistrācijas iesnieguma iesniedzējam to, ka
ražotājs nodrošina visu sēriju atbilstību paraugam un ka ražotājs
bez reģistrācijas iesnieguma iesniedzēja ziņas nemaina ražošanas
procesu vai specifikācijas. Kompetentajām iestādēm iesniedz
dokumentus un ziņas, ko pievieno pieteikumam attiecībā uz šādām
maiņām;
9.3. ziņās un dokumentos, ko
pievieno iesniegumam reģistrācijas apliecības saņemšanai
atbilstoši šo noteikumu 13.9. un 13.10.apakšpunktā un 18., 19.,
20. un 21.punktā noteiktajām prasībām, iekļauj visu izmantoto
komponentu kvalitātes kontroles testu rezultātus, ietverot sēriju
analīzi attiecībā uz aktīvajām vielām. Rezultātus iesniedz
saskaņā ar turpmāk minētiem nosacījumiem;
9.4. izejmateriāli, kas uzskaitīti
farmakopejās:
9.4.1. Eiropas Farmakopejas
monogrāfijas attiecas uz visām tajā sastopamajām vielām;
9.4.2. par citām vielām katra
dalībvalsts var pieprasīt, lai attiecībā uz tās teritorijā
ražotajām zālēm tiktu ievērota dalībvalsts nacionālā
farmakopeja;
9.4.3. komponenti, kas atbilst
Eiropas Farmakopejas prasībām vai kādas dalībvalsts farmakopejas
prasībām, uzskata par pietiekamā mērā atbilstošiem šo noteikumu
13.9.apakšpunkta prasībām. Šajā gadījumā analītisko metožu
aprakstu var aizstāt ar sīki izstrādātu atsauci uz minēto
farmakopeju;
9.4.4. ja Eiropas Farmakopejā vai
dalībvalsts farmakopejā minētais izejmateriāls ir pagatavots ar
metodi, kas var atstāt piemaisījumus, ko nekontrolē farmakopejas
monogrāfija, norāda šos piemaisījumus un to maksimālās pielaides
robežas un apraksta piemērotu testēšanas procedūru;
9.4.5. krāsvielas visos gadījumos
atbilst šo noteikumu 4.pielikuma prasībām;
9.4.6. regulārie testi, ko veic
katrai izejmateriālu sērijai, atbilst testiem, kas norādīti
iesniegumā reģistrācijas apliecības saņemšanai. Ja izmanto
testus, kas nav minēti farmakopejā, tad sniedz pierādījumus, ka
izejmateriāli atbilst farmakopejā noteiktajām kvalitātes
prasībām;
9.4.7. ja specifikācija, kas
ierakstīta Eiropas Farmakopejā vai dalībvalsts farmakopejā, nav
pietiekama, lai nodrošinātu vielas kvalitāti, Zāļu valsts
aģentūra var pieprasīt no reģistrācijas atļaujas turētāja
specifikācijas, kas nodrošina vielas kvalitāti;
9.4.8. Zāļu valsts aģentūra
informē iestādes, kas atbildīgas par attiecīgo farmakopeju.
Reģistrācijas atļaujas turētājs sniedz šo farmakopeju
pārzinošajām iestādēm sīkas ziņas par varbūtējo nepilnību un par
papildus izmantotajām specifikācijām;
9.4.9. ja izejmateriāls nav
aprakstīts Eiropas Farmakopejā un Latvijas farmakopejā, var
akceptēt atbilstību monogrāfijai kādas trešās valsts farmakopejā.
Šādos gadījumos reģistrācijas iesnieguma iesniedzējs sagādā
monogrāfijas kopiju, kam vajadzības gadījumā pievienots
monogrāfijā esošo testēšanas procedūru apstiprinājums un, ja
nepieciešams, tulkojums;
9.5. Izejmateriālu komponentus,
kas nav uzskaitīti nevienā farmakopejā, apraksta monogrāfijas
veidā, iedalot šādi:
9.5.1. vielas nosaukumam, kas
atbilst šī pielikuma 6.2.apakšpunkta prasībām, pievieno
komerciālos vai zinātniskos sinonīmus;
9.5.2. vielas definīcijai, kas
izklāstīta tādā formā, kādu lieto Eiropas Farmakopejā, pievieno
nepieciešamo paskaidrojumu, jo īpaši tādu, kas attiecīgā gadījumā
skar molekulāro struktūru. Tai pievieno pienācīgu sintēzes
paņēmiena aprakstu. Ja vielas var aprakstīt tikai pēc to
ražošanas metodes, tad apraksts ir pietiekami precīzi izklāstīts,
lai raksturotu tādu vielu, kura ir konstanta gan sastāva, gan
iedarbības ziņā;
9.5.3. identifikācijas metodes var
būt aprakstītas kā pilnās metodes, ko izmanto vielas ražošanai,
un kā regulāri veicami testi;
9.5.4. tīrības testi aprakstīti
saistībā ar to kopējo daudzumu, ko veido prognozētie
piemaisījumi, īpaši tādi, kam var būt kaitīgas sekas, un, ja
vajadzīgs, tie, kas, ņemot vērā vielu salikumu, uz kuru attiecas
pieteikums, varētu negatīvi ietekmēt zāļu stabilitāti vai
izkropļot analīzes rezultātus;
9.5.5. attiecībā uz kompleksām
augu vai dzīvnieku izcelsmes vielām norāda atšķirību starp
gadījumu, kad daudzveidīgas farmakoloģiskās iedarbības dēļ
vajadzīga galveno sastāvdaļu ķīmiska, fizikāla vai bioloģiska
kontrole, un gadījumu, kad vielās ir viena vai vairākas galveno
vielu grupas ar līdzīgu darbību, kam var akceptēt vispārēju
novērtējuma metodi;
9.5.6. ja izmanto dzīvnieku
izcelsmes materiālus, apraksta pasākumus, kas nodrošina iespējamo
patogēnu novēršanu;
9.5.7. norāda visus īpašos
piesardzības pasākumus, kas var būt vajadzīgi izejmateriāla
uzglabāšanai, un vajadzības gadījumā– maksimālo līdz atkārtotai
pārbaudei pieļaujamo uzglabāšanas laikposmu;
9.6. fizikāli ķīmiskās īpašības,
kas var ietekmēt bioloģisko pieejamību:
9.6.1. šādu informāciju par
aktīvajām vielām (neatkarīgi no tā, vai tās ir uzskaitītas
farmakopejās) sniedz kā aktīvo vielu vispārējā apraksta
sastāvdaļu, ja no tā atkarīga zāļu bioloģiskā pieejamība:
9.6.1.1. kristāliskā forma un
šķīdības koeficienti;
9.6.1.2. daļiņu lielums, attiecīgā
gadījumā pēc pulverizēšanas;
9.6.1.3. solvācijas pakāpe;
9.6.1.4. eļļas vai ūdens
sašķelšanās koeficients;
9.6.2. 9.6.1.1., 9.6.1.2. un
9.6.1.3.apakšpunktu nepiemēro vielām, ko lieto vienīgi
šķīdumā;
9.7. ja veterināro zāļu ražošanā
izmanto tādus resursu avotus kā mikroorganismi, augu vai
dzīvnieku izcelsmes audi, šūnas vai šķidrumi (ietverot asinis),
kas iegūti no dzīvniekiem vai cilvēkiem, vai biotehnoloģiskas
šūnu struktūras, tad apraksta izejmateriālu izcelsmi un vēsturi
un pamato ar dokumentiem;
9.8. izejmateriāla aprakstā
iekļauj ražošanas stratēģiju, attīrīšanas vai inaktivācijas
metodes kopā ar to apstiprinājumu un visas procedūrās attiecībā
uz kontroli ražošanas gaitā ar uzdevumu nodrošināt visu
galaprodukta sēriju atbilstību paraugam;
9.9. ja izmanto šūnu bankas, tad
jāatspoguļo tas, ka šūnu raksturlielumi saglabājas nemainīgi
pirms un pēc ražošanā lietotās pasāžas;
9.10. kultūras materiālus, šūnu
bankas, serumu un citu bioloģiskas izcelsmes materiālu
apkopojumus un, ja iespējams, resursu avotus, no kuriem tie
iegūti, pārbauda attiecībā uz papildu slimības izraisītājiem;
9.11. ja nav iespējams izvairīties
no iespējamas patogēno izraisītāju klātbūtnes, šo materiālu
izmanto tikai tādā gadījumā, ja tālākā apstrāde nodrošina
patogēno izraisītāju likvidēšanu un/vai inaktivāciju un tas tiek
apstiprināts.
10. Īpaši pasākumi, lai novērstu
dzīvnieku sūkļveida encefalopātijas pārnešanu– reģistrācijas
iesnieguma iesniedzējs uzrāda, ka veterinārās zāles ir ražotas
saskaņā ar Komisijas norādījumiem par riska samazināšanu līdz
minimumam attiecībā uz iespēju veterinārām zālēm pārnēsāt
dzīvnieku sūkļveida encefalopātijas izraisītājus, ko Eiropas
Komisija publicējusi Eiropas Kopienas Zāļu reglamentācijas
noteikumu 7.sējumā, un to jaunākajām redakcijām, kuru tulkojums
pieejams Zemkopības ministrijas mājas lapā.
11. Ražošanas starpposmos veiktie
kontroles testi:
11.1. ziņās un dokumentos, ko
pievieno reģistrācijas iesniegumam atbilstoši šo noteikumu 13.9.
un 13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām, iekļauj ziņas par izstrādājuma kontroles testiem, ko
iespējams veikt kādā ražošanas starpposmā, lai nodrošinātu
tehnisko parametru un ražošanas procesa atbilstību;
11.2. šiem testiem ir būtiska
nozīme, lai pārbaudītu zāļu atbilstību formulai, ja reģistrācijas
iesnieguma iesniedzējs izņēmuma kārtā piedāvā gatavā produkta
testēšanai tādu analīzes metodi, kurā netiek iekļauts visu aktīvo
vielu (vai visu palīgvielas sastāvdaļu, uz kuriem attiecas tās
pašas prasības kā uz aktīvajām vielām) vērtējums;
11.3. tas pats attiecas uz
gadījumiem, kad galaprodukta kvalitātes kontrole ir atkarīga no
kontroles ražošanas gaitā, jo īpaši, ja attiecīgo vielu būtiski
nosaka tās ražošanas metode.
12. Gatavās produkcijas
kontrole:
12.1. veicot gatavās produkcijas
kontroli, ar gatavās produkcijas sēriju saprot kādas zāļu formas
visas vienības, kas izgatavotas no viena un tā paša sākotnējā
materiāla daudzuma un kam veikta vienu un to pašu ražošanas
un/vai sterilizācijas operāciju virkne, vai arī nepārtrauktā
ražošanas procesa gadījumā– visas noteiktā laika posmā saražotās
vienības;
12.2. reģistrācijas iesniegumā
uzskaita tos testus, ko regulāri veic katrai gatavās produkcijas
sērijai. Nosaka to testu biežumu, kurus neveic regulāri. Norāda
izlaides robežas;
12.3. ziņās un dokumentos, ko
pievieno reģistrācijas iesniegumam atbilstoši šo noteikumu 13.9.
un 13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām, iekļauj ziņas par kontroli, ko veic, izlaižot gatavo
produkciju. Tos iesniedz saskaņā ar turpmāk minētiem
nosacījumiem;
12.4. Eiropas Farmakopejas
vispārējo monogrāfiju vai, ja tādu nav, dalībvalsts farmakopejas
monogrāfiju nosacījumi attiecas uz visiem tur definētajiem
izstrādājumiem;
12.5. ja lieto citas testēšanas
procedūras un robežlielumu, nevis tos, kas ierakstīti Eiropas
Farmakopejas vispārējās monogrāfijās, tad sniedz pierādījums par
to, ka gatavais izstrādājums tā testēšanas gadījumā saskaņā ar šo
monogrāfiju atbilst šādas farmakopejas kvalitātes prasībām
attiecībā uz konkrēto zāļu formu;
12.6. gatavā izstrādājuma
vispārīgās īpašības:
12.6.1. gatavā izstrādājuma
testēšanā iekļauj noteiktus testus par izstrādājuma vispārīgām
īpašībām. Šie testi attiecas uz vidējā svara un maksimālās
novirzes kontroli, uz mehāniskiem, fizikāliem vai
mikrobioloģiskiem testiem, organoleptiskajām īpašībām, fizikālām
īpašībām, tādām kā blīvums, pH, atstarošanas indekss utt.
Reģistrācijas iesnieguma iesniedzējs katrā atsevišķā gadījumā
nosaka standartus un pielaides robežas katrai no šīm
īpašībām;
12.6.2. precīzi un sīki apraksta
testēšanas apstākļus, attiecīgā gadījumā, izmantotās iekārtas vai
aparatūru un standartus, ja tie nav sniegti Eiropas Farmakopejā.
Tas pats attiecas uz gadījumiem, kad šo farmakopeju noteiktās
metodes nav piemērojamas;
12.6.3. bez tam cieto zāļu formām,
kas ievadāmas caur muti, pārbauda in vitro aktīvās
vielas vai vielu atbrīvošanās un izšķīšanas ātrumu. Zāļu valsts
aģentūra ir tiesīga pieprasīt šādus pētījumus veikt arī
gadījumos, ja ievadīšanas veids ir cits;
12.7. aktīvās vielas (aktīvo
vielu) identifikācija un novērtējums:
12.7.1. aktīvās vielas (aktīvo
vielu) identifikāciju un novērtējumu veic produkcijas partijas
raksturīgā paraugā vai arī virknē dozējuma vienību, ko analizē
individuāli;
12.7.2. ja nav attiecīga
pamatojuma, tad gatavās produkcijas aktīvās vielas satura
maksimāli pieļaujamā novirze nepārsniedz 5 % ražošanas laikā;
12.7.3. balstoties uz stabilitātes
testiem, ražotājs ierosina un pamato gatavās produkcijas aktīvās
vielas satura maksimāli pieļaujamās pielaides robežas līdz
ierosinātā derīguma termiņa beigām;
12.7.4. noteiktos izņēmuma
gadījumos ar īpaši sarežģītiem maisījumiem, kad ļoti
daudzskaitlīgu vai ļoti mazos daudzumos pārstāvētu aktīvo vielu
novērtēšanai būtu vajadzīgs sarežģīts pētījums, ko grūti veikt
attiecībā uz katru produkcijas sēriju, gatavajā produkcijā var
neveikt vienas vai vairāku aktīvo vielu novērtēšanu ar skaidru
nosacījumu, ka šos novērtējumus veic ražošanas procesa
starpposmos. Šādu atkāpi prasībās neattiecina uz šo vielu
raksturojumu. Šo vienkāršoto metodi papildina ar kvantitatīvā
izvērtējuma metodi, kas ļauj Zāļu valsts aģentūrai pārliecināties
par tirgū laistu zāļu atbilstību apstiprinātajai
specifikācijai;
12.7.5. ja fizikāli ķīmiskās
metodes nesniedz adekvātu informāciju par izstrādājuma kvalitāti,
tad ir obligāti veicama in vivo vai in vitro
bioloģiska novērtēšana. Šāda novērtēšana pēc iespējas ietver sevī
atsauces materiālus un statistikas analīzi, kas ļauj aprēķināt
ticamības pakāpi. Ja nevar veikt gatavās produkcijas testus, tos
var veikt pēc iespējas vēlākā ražošanas procesa starpposmā;
12.7.6. ja šī pielikuma 8.punktā
sniegtās ziņas norāda uz to, ka zāļu ražošanā lieto ievērojamu
aktīvās vielas devas palielinājumu, tad gatavās produkcijas
kontroles testu aprakstā attiecīgā gadījumā iekļauj to pārmaiņu
ķīmisko un, ja vajadzīgs, toksikoloģiski farmakoloģisko pētījumu,
kam pakļauta šī viela, un pēc iespējas noārdīšanās produktu
raksturojumu un/vai novērtējumu;
12.8. palīgvielas sastāvdaļu
identifikācija un novērtējums:
12.8.1. ciktāl nepieciešams,
palīgvielas sastāvdaļām veic vismaz identifikācijas testus;
12.8.2. testēšanas procedūra, ko
piedāvā krāsvielu identificēšanai, ļauj pārliecināties, ka šādas
vielas ir uzskaitītas šo noteikumu 4.pielikumā;
12.8.3. attiecībā uz konservantiem
obligāti veic augšējās un apakšējās robežas testu, un attiecībā
uz jebkuru citu papildvielas sastāvdaļu, kas varētu nelabvēlīgi
ietekmēt fizioloģiskās funkcijas– augšējās robežas testu.
Augšējās un apakšējās robežas testu obligāti veic attiecībā uz
papildvielu, kas var ietekmēt aktīvās vielas bioloģisko
pieejamību, ja vien bioloģisko pieejamību nenodrošina citi
attiecīgi testi;
12.9. drošuma testi– līdztekus
toksikoloģiski farmakoloģiskajiem testiem, kas iesniegti
reģistrācijas iesniegumā, analīzes datos iekļauj ziņas par
drošuma testiem, piemēram, attiecībā uz sterilitāti,
bakteriālajiem endotoksīniem, pirogēniem un vietējo panesamību
dzīvniekiem, ja šādus testus veic regulāri, nodrošinot
izstrādājuma kvalitāti.
13. Stabilitātes tests. Ziņas un
dokumentus, ko pievieno iesniegumam reģistrācijas apliecības
saņemšanai atbilstoši šo noteikumu 13.9. un 13.10.apakšpunktā
noteiktajām prasībām, iesniedz saskaņā ar šādām prasībām:
13.1. iesniedz aprakstu par
pētījumu, kur ir noteikts reģistrācijas iesnieguma iesniedzēja
piedāvātais derīguma termiņš, ieteiktie uzglabāšanas nosacījumi
un specifikācijas derīguma termiņa beigās;
13.2. dzīvnieku ārstnieciskās
barības premiksu gadījumā, ja nepieciešams, sniedz informāciju
arī par tādas ārstnieciskās dzīvnieku barības derīguma termiņu,
kas ražota no šiem premiksiem saskaņā ar ieteikto lietošanas
pamācību;
13.3. ja paredzēts gatavās zāles
atjaunot, nepieciešamas sīkas ziņas attiecībā uz atjaunoto zāļu
ieteicamo derīguma termiņu, kam pievieno atbilstošos datus par
stabilitāti;
13.4. attiecībā uz vairāku devu
iepakojumu iesniedz datus par stabilitāti, kas pamato iepakojuma
derīguma termiņu pēc tā pirmreizējās atvēršanas;
13.5. ja gatavais izstrādājums
varētu radīt noārdīšanās produktus, reģistrācijas iesnieguma
iesniedzējs to uzrāda un norāda uz raksturošanas metodēm un
testēšanas procedūrām;
13.6. slēdzienā iekļauj analīžu
rezultātus, kas pamato piedāvāto derīguma termiņu ieteiktajos
uzglabāšanas apstākļos un gatavā produkta specifikācijas gatavā
produkta derīguma beigās šajos ieteiktajos uzglabāšanas
apstākļos;
13.7. norāda maksimāli pieļaujamo
noārdīšanās produktu līmeni zāļu derīguma termiņa beigās;
13.8. iesniedz pētījumu par zāļu
un trauka savstarpējo mijiedarbību, ja vien šādas mijiedarbības
risks tiek uzskatīts par iespējamu, jo īpaši, ja tas skar
injicējamus pagatavojumus vai aerosolus iekšķīgai lietošanai.
III. Testēšana
attiecībā uz drošumu un atliekvielām
14. Ziņas un dokumentus, ko
pievieno reģistrācijas iesniegumam atbilstoši 13.9. un
13.10.apakšpunktā noteiktajām prasībām, iesniedz saskaņā ar šajā
nodaļā noteiktām prasībām.
15. Dalībvalstis nodrošina, ka
testus veic saskaņā ar labas laboratoriju prakses principiem
atbilstoši normatīvajiem aktiem par prasībām laboratoriju darba
kvalitātei un laboratoriju inspicēšanai.
16. Drošuma testēšana:
16.1. dokumentos attiecībā uz
drošumu atspoguļo:
16.1.1. veterināro zāļu iespējamo
toksiskumu un jebkuras bīstamas vai nevēlamas blakusparādības,
kas iespējamas apstākļos, kādos veterinārās zāles lieto
dzīvniekiem. Iespējamo toksiskumu un blakusparādības izvērtē
saistībā ar attiecīgā patoloģiskā stāvokļa smaguma pakāpi;
16.1.2. iespējamo kaitīgo ietekmi
uz cilvēkiem, ko var izraisīt veterināro zāļu atliekvielas
pārtikas produktos, kas iegūti no apstrādātajiem dzīvniekiem, un
to, kādus sarežģījumus šīs atliekvielas var radīt pārtikas
produktu rūpnieciskajā pārstrādē;
16.1.3. iespējamo risku, kas var
rasties, ja cilvēks ir pakļauts veterināro zāļu iedarbībai,
piemēram, tā ievadīšanas laikā dzīvniekam;
16.1.4. iespējamo vides
apdraudējumu, ko var izraisīt veterināro zāļu lietošana;
16.2. visi rezultāti ir ticami un
vispārēji derīgi. Eksperimenta metodes izveidē un rezultātu
novērtēšanā attiecīgā gadījumā lieto matemātikas un statistikas
metodes. Turklāt klīnicistus (ekspertus, klīnisko pētījumu
veicējus) informē par izstrādājuma jaunāko terapeitisko
potenciālu un par risku, kas saistīts ar tā lietošanu;
16.3. dažos gadījumos var būt
nepieciešams testēt izejas savienojumu metabolītus, ja tie parāda
attiecīgās atliekvielas;
16.4. attiecībā uz tādu
palīgvielu, ko izmanto farmācijā pirmo reizi, rīkojas tāpat kā
aktīvas vielas gadījumā.
17. Farmakoloģija:
17.1. farmakoloģiskiem pētījumiem
ir fundamentāla nozīme, noskaidrojot mehānismus, kādos zāles gūst
savu terapeitisko iedarbību. Šī pielikuma IV daļā iekļauj
farmakoloģiskos pētījumus, kas veikti ar izmēģinājuma dzīvniekiem
un ar mērķa sugām;
17.2. farmakoloģiskie pētījumi
palīdz izprast parādības toksikoloģijā. Ja zāles gūst
farmakoloģisku iedarbību bez toksiskas reakcijas vai arī pie tik
zemām devām, kas nevar izraisīt toksisku reakciju, šo
farmakoloģisko iedarbību ņem vērā, novērtējot zāļu drošumu;
17.3. dokumentiem par zāļu drošumu
papildus iesniedz sīkas ziņas par farmakoloģiskajiem pētījumiem,
kas veikti ar laboratorijas dzīvniekiem, un visa attiecīgā
informācija, kas gūta, veicot klīniskos pētījumus ar mērķa
dzīvniekiem.
18. Toksikoloģija:
18.1. Vienreizējas devas
toksiskums. Pētījumus par vienreizējas devas toksiskumu izmanto,
lai noskaidrotu:
18.1.1. iespējamās sekas akūtas
pārdozēšanas gadījumā mērķa sugai;
18.1.2. iespējamās sekas gadījumā,
kad to nejauši kļūmīgi ievada cilvēkam;
18.1.3. devas, ko lietderīgi
izmanto pētījumiem par atkārtoto devu;
18.2. pētījumi par vienreizējas
devas toksiskumu atklāj vielas akūto toksisko iedarbību, kā arī
tās iestāšanās un izzušanas laiku;
18.3. šajos pētījumos izmanto ne
mazāk kā divas zīdītāju sugas. Vienu zīdītāju sugu iespējams
aizstāt ar dzīvnieku sugu, kam zāles paredzētas. Parasti pēta ne
mazāk kā divus dažādus ievadīšanas veidus. Viens no tiem var būt
tas ievadīšanas veids, ko iesaka attiecībā uz mērķa sugu, vai
līdzīgs tam. Ja ir paredzama lietotāja būtiska saskarsme ar
zālēm, piemēram, inhalācijas vai ādas kontakta veidā, tad izpēta
šos lietošanas veidus;
18.4. lai mazinātu izmēģinājumos
iesaistīto dzīvnieku skaitu un to ciešanas, pastāvīgi izstrādā
jaunus protokolus vienreizējas devas toksiskuma testēšanai. Tiek
akceptēti pētījumi, kas veikti saskaņā ar šīm jaunajām
procedūrām, ja tās ir pienācīgi apstiprinātas, kā arī pētījumi,
kas veikti saskaņā ar pastāvošām starptautiski atzītām
pamatnostādnēm;
18.5. atkārtotas devas
toksiskums:
18.5.1. atkārtotas devas
toksiskuma testi ir paredzēti, lai atklātu jebkādas fizioloģiskās
un/vai patoloģiskās pārmaiņas, ko rada pētāmās aktīvās vielas vai
aktīvo vielu kombinācijas atkārtota lietošana, un lai noteiktu
šīs pārmaiņas atkarībā no devas dozējuma;
18.5.2. ja zāles vai vielas
paredzēts izmantot neproduktīviem dzīvniekiem, veic atkārtotas
devas toksiskuma pētījumu vienai izmēģinājumu dzīvnieku sugai. Šā
pētījuma vietā var veikt pētījumu ar mērķa dzīvnieku. Ievadīšanas
veidu un biežumu, kā arī pētījuma ilgumu izvēlas, ievērojot
paredzētos klīniskos lietošanas apstākļus. Pētnieks argumentē
izmēģinājumu apjoma un ilguma, kā arī devu izvēli;
18.5.3. ja zāles vai vielas
paredzēts izmantot produktīviem dzīvniekiem, pētījumu veic ne
mazāk kā ar divām dzīvnieku sugām, no kurām viena nav grauzēji.
Pētnieks argumentē dzīvnieku sugu izvēli, ņemot vērā pieejamās
zināšanas par produkta metabolismu dzīvnieku un cilvēka
organismā. Testa vielu ievada caur muti. Testa ilgums ir vismaz
90 dienas. Pētnieks skaidri nosaka un argumentē lietošanas veidu
un biežumu un izmēģinājumu ilgumu;
18.5.4. maksimāli pieļaujamo devu
izvēlas, lai atklātu iespējamu kaitīgu iedarbību. Zemākais devas
līmenis nedrīkst uzrādīt nekādas toksiskuma pazīmes;
18.5.5. toksiskās iedarbības
novērtējums balstās uz novērojumiem par uzvedību, augšanu,
hematoloģiskiem un fizioloģiskiem testiem, jo īpaši attiecībā uz
izvadorgāniem, kā arī sekcijas rezultātiem un pievienotajiem
histoloģijas datiem. Katras testu grupas izvēle un diapazons ir
atkarīgs no izmantotās dzīvnieku sugas un no jaunākajām zinātnes
atziņām;
18.5.6. ja zināmas vielas, kas
izpētītas saskaņā ar šo noteikumu prasībām, savieno jaunās
kombinācijās, pētnieks var modificēt atkārtotas devas testus,
pamatojot modifikāciju, ja vien toksiskuma testi neliecina par
potencēšanu vai jaunu toksisku iedarbību;
18.6. tolerance mērķa sugā– sniedz
sīkas ziņas par jebkādām nepanesības pazīmēm, kas novērotas,
veicot pētījumu mērķa sugai saskaņā ar šī pielikuma 28.punkta
prasībām. Norāda:
18.6.1. attiecīgo pētījumu,
18.6.2. devas, pie kurām vērojama
nepanesība,
18.6.3. attiecīgās sugas un
šķirnes, kam novērojama nepanesība,
28.6.4. sīki izklāstīta
informācija par jebkādām negaidītām fizioloģiskām pārmaiņām,
veicot attiecīgu pētījumu;
18.7. reproduktīvais toksiskums,
ietverot teratogēno toksiskumu– pētījums par ietekmi uz
reproduktīvo jomu:
18.7.1. pētījuma uzdevums ir
noteikt iespējamo vīrišķās vai sievišķās reproduktīvās funkcijas
pasliktināšanos vai iespējamu kaitīgu iedarbību uz pēcnācējiem,
ko izraisa zāļu vai pētāmās vielas izmantošana;
18.7.2. vielām vai zālēm, ko
paredzēts izmantot produktīviem dzīvniekiem, pētījumu par ietekmi
uz reproduktīvo jomu veic kā divu paaudžu pētījumu ne mazāk kā
vienai dzīvnieku sugai, parasti– grauzējiem. Pētāmo vielu vai
zāles ievada vīriešu un sieviešu kārtas dzīvniekiem atbilstošā
termiņā pirms pārošanas. Pētāmo vielu vai zāles turpina ievadīt
līdz pat F2 paaudzes atšķiršanai no zīdītājas. Izmanto ne mazāk
kā trīs devu lielumus. Maksimāli pieļaujamā deva būtu jāizvēlas,
lai atklātu iespējamu kaitīgu iedarbību. Zemākais devas līmenis
neuzrāda toksiskuma pazīmes;
18.7.3. novērtējumu par ietekmi uz
reproduktīvo jomu balsta uz auglību, grūsnību un ciltsmātes
izturēšanos; F1 pēcnācēju zīšanu, augšanu un attīstību no
apaugļošanās brīža līdz nobriešanai; F2 pēcnācēju attīstību– līdz
atšķiršanai no zīdītājas;
18.7.4. pētījums par toksisko
iedarbību uz embriju vai augli, ietverot teratogēno
toksiskumu;
18.7.5. vielām vai zālēm, ko
paredzēts lietot produktīviem dzīvniekiem, veic pētījumus par
toksisko iedarbību uz embriju vai augli, ietverot teratogēno
toksiskumu. Šos pētījumus veic vismaz divām zīdītāju sugām,
parasti– ar kādu no grauzējiem un ar trušiem. Sīku ziņu izklāsts
par testu (dzīvnieku skaits, devas, ievadīšanas laiki un
kritēriji rezultātu izvērtēšanai) ir atkarīgs no zinātnes
attīstības līmeņa reģistrācijas iesnieguma iesniegšanas brīdī un
no statistiskā svarīguma pakāpes, ko gaida no šiem rezultātiem.
Pētījumu ar grauzējiem var apvienot ar pētījumu par ietekmi uz
reproduktīvo funkciju;
18.7.6. vielām vai zālēm, ko nav
paredzēts lietot produktīvajiem dzīvniekiem, pētījumu par
toksisko iedarbību uz embriju vai augli, ietverot teratogēno
toksiskumu, veic vismaz ar vienu sugu, kas var būt mērķa suga, ja
zāles paredzēts lietot iespējamiem vaislas dzīvniekiem;
18.8. mutagenitāte:
18.8.1. mutagenitātes testus
paredz, lai novērtētu vielu spēju izraisīt transmisīvas pārmaiņas
šūnu ģenētiskajā materiālā;
18.8.2. jaunas vielas, ko
paredzēts lietot veterinārajās zālēs, izvērtē attiecībā uz to
mutagēnajām īpašībām;
18.8.3. testu skaita un veida
izvēle un rezultātu novērtēšanas kritēriju izvēle ir atbilstoša
zinātnes attīstības līmenim laikā, kad iesniedz reģistrācijas
iesniegumu;
18.9. kancerogenitāte:
18.9.1. ilgstošus pētījumus
attiecībā uz kancerogenitāti dzīvniekiem veic vielām, kuru
iedarbībai pakļauti cilvēki. Šīs vielas:
18.9.1.1. pēc ķīmiskā sastāva
analoģiski līdzīgas zināmajiem kancerogēniem;
18.9.1.2. mutagenitātes testos
uzrāda rezultātus par kancerogēnas iedarbības iespējamību;
18.9.1.3. toksiskuma testēšanas
laikā uzrāda pazīmes par kancerogēnās iedarbības iespējamību;
18.9.2. veicot pētījumus attiecībā
uz kancerogenitāti un izvērtējot to rezultātus, ņem vērā zinātnes
attīstības līmeni pieteikuma iesniegšanas laikā;
18.10. izņēmumi– ja zāles
paredzēts lietot lokāli, pētījumu veic par sistēmisko absorbciju
dzīvnieku mērķa sugā. Ja pierāda, ka sistēmiskā absorbcija ir
nenozīmīga, tad var neveikt atkārtotas devas toksiskuma testu,
testus attiecībā uz reproduktīvo toksiskumu un testus attiecībā
uz kancerogenitāti, ja vien nav izrādījies, ka:
18.10.1. saskaņā ar noteiktajiem
lietošanas nosacījumiem dzīvnieks, iespējams, uzņems zāles caur
muti;
18.10.2. zāļu daļiņas var iekļūt
pārtikas produktos, ko iegūst no apstrādātā dzīvnieka (piena
dziedzeros ievadāmi pagatavojumi).
19. Citas prasības:
19.1. imūntoksiskums:
19.1.1. ja dzīvniekiem veiktajos
atkārtotās devas pētījumos novērotā iedarbība ietver sevī
specifiskas pārmaiņas attiecībā uz limfas orgānu svaru un/vai
histoloģiju un šūnu veidošanās pārmaiņas limfaudos, kaulu
smadzenēs vai perifērajos leikocītos, pētnieks lemj par
nepieciešamību veikt papildu pētījumus par zāļu ietekmi uz
imūnsistēmu;
19.1.2. veicot šos pētījumus un
izvērtējot to rezultātus, ņem vērā zinātnes attīstības līmeni
pieteikuma iesniegšanas laikā;
19.2. atliekvielu mikrobioloģiskās
īpašības:
19.2.1. iespējamā iedarbība uz
cilvēka zarnu floru– mikrobioloģisko risku, ko cilvēka zarnu
mikroflorai rada savienojumi ar pretmikrobu īpašībām, pēta
saskaņā ar zinātnes attīstības līmeni pieteikuma iesniegšanas
laikā;
19.2.2. iespējamā iedarbība uz
pārtikas rūpnieciskajā pārstrādē izmantojamiem organismiem –
atsevišķos gadījumos var veikt testus, lai noteiktu, vai
atliekvielas izraisa sarežģījumus attiecībā uz tehnoloģiskajiem
procesiem pārtikas rūpnieciskajā pārstrādē;
19.3. novērojumi attiecībā uz
cilvēka organismu– sniedz ziņas par to, vai veterināro zāļu
komponenti tiek lietoti kā zāles, ārstējot cilvēkus. Ja šāds
fakts pastāv, tad sniedz pārskatu par visām novērotajām ietekmēm
(ietverot blakusparādības) attiecībā uz cilvēkiem un par šo
ietekmju iemeslu, lai būtu iespējams veikt veterināro zāļu
novērtēšanu, ņemot vērā pētījumu rezultātu bibliogrāfiju. Ja
veterināro zāļu komponentus nelieto vai tos vairs nelieto kā
zāles cilvēku ārstēšanai, tad norāda iemeslus.
20. Ekotoksiskums:
20.1. veterināro zāļu
ekotoksiskuma pētījuma uzdevums ir novērtēt zāļu lietošanas
iespējamo kaitīgo ietekmi uz vidi un noteikt visus vajadzīgos
piesardzības pasākumus šā riska samazināšanai;
20.2. ekotoksiskuma novērtējums ir
obligāts iesniegumam reģistrācijas apliecības saņemšanai, izņemot
tādus iesniegumus, kas iesniegti saskaņā ar šo noteikumu 13.9. un
13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām;
20.3. novērtējumu veic divos
posmos. Pirmajā posmā pētnieks novērtē apjomu, kādā ir iespējama
zāļu, to aktīvo vielu vai attiecīgo metabolītu ietekme uz vidi,
ņemot vērā:
20.3.1. mērķa sugu un piedāvāto
lietošanas modeli (piemēram, zāļu masveida lietošana dzīvniekiem
vai zāļu lietošana atsevišķam dzīvniekam);
20.3.2. lietošanas veidu, jo
īpaši– apjomu, kādā zāles iekļūs tieši vides sistēmās;
20.3.3. zāļu, to aktīvo vielu vai
metabolītu iespējamo izdalīšanos no apstrādāto dzīvnieku
organisma vidē un noturību šādos izdalījumos;
20.3.4. neizmantoto zāļu vai no
tām radušos atkritumu likvidēšana;
20.4. otrajā posmā, ņemot vērā
zāļu ietekmes uz vidi apjomu un pieejamo informāciju par tā
sastāva fizikālajām/ķīmiskajām, farmakoloģiskajām un/vai
toksikoloģiskajām īpašībām, kas iegūta, veicot citus testus un
izmēģinājumus, kas noteikti šajos noteikumos, pētnieks lemj par
nepieciešamību veikt tālākus pētījumus par zāļu ietekmi uz
konkrētām ekosistēmām;
20.5. var būt vajadzīgi tālāki
pētījumi attiecībā uz:
20.5.1. saglabāšanos un darbību
augsnē;
20.5.2. saglabāšanos un darbību
ūdenī un gaisā;
20.5.3. iedarbību uz ūdens
organismiem;
20.5.4. iedarbību uz citiem blakus
sugu organismiem;
20.6. šos tālākos pētījumus veic
saskaņā ar testa protokoliem, kas noteikti normatīvajos aktos par
laboratoriskajām metodēm ķīmisko vielu un ķīmisko produktu
fizikālo, ķīmisko, toksikoloģisko vai ekotoksikoloģisko īpašību
noteikšanai. Ja testa protokolos nav skaidri norādīts gala
termiņš, tālākos pētījumus veic saskaņā ar citiem starptautiski
atzītiem protokoliem par veterināro zāļu un/vai aktīvajām vielām
un/vai attiecīgi izdalītajiem metabolītiem. Testu skaita un veida
izvēle un rezultātu novērtēšanas kritēriju izvēle ir atbilstoša
zinātnes attīstības līmenim laikā, kad iesniedz pieteikumu.
21. Drošuma testu dokumentācijā kā
jebkurā zinātniskā darbā iekļauj šādas ziņas:
21.1. ievadu, kurā definē
aplūkojamo objektu un kuram pievienotas noderīgas bibliogrāfiskās
atsauces;
21.2. aplūkojamās vielas sīki
izklāstīta identifikācija, ietverot:
21.2.1. starptautisko nepatentēto
nosaukumu (INN);
21.2.2. Starptautiskās teorētiskās
un praktiskās ķīmijas apvienības nomenklatūras (IUPAC)
nosaukumu;
21.2.3. Ķīmijas analītisko apskatu
indeksa (Chemical Abstract Service– CAS) numuru;
21.2.4. terapeitisko un
farmakoloģisko klasifikāciju;
21.2.5. sinonīmus un
saīsinājumus;
21.2.6. struktūrformulu;
21.2.7. molekulāro formulu;
21.2.8. molekulmasu;
21.2.9. piemaisījumu līmeni;
21.2.10. piemaisījumu kvalitatīvo
un kvantitatīvo sastāvu;
21.2.11. fizikālo īpašību
aprakstu;
21.2.12. kušanas punktu;
21.2.13. viršanas punktu;
21.2.14. tvaika spiedienu;
21.2.15. šķīdību ūdenī un
organiskos šķīdinātājos, kas izteikta g/l, norādot
temperatūru;
21.2.16. blīvumu;
21.2.17. refrakcijas, rotācijas
spektru utt.;
21.3. sīku eksperimenta protokolu,
kurā sniedz pamatojumu, ja izlaisti kādi no iepriekš minētiem
testiem, izmantoto metožu, aparatūras un materiālu aprakstu,
sīkas ziņas par dzīvnieku sugu, šķirni vai līniju, ziņas par
dzīvnieku iegūšanu, skaitu, izmitināšanas apstākļiem un barošanas
nosacījumiem, inter alia nosakot, vai dzīvnieki ir brīvi
no specifiskiem patogēniem (SPF);
21.4. iegūtos rezultātus
neatkarīgi no tā, vai tie ir labvēlīgi vai nelabvēlīgi.
Sākotnējos datus apraksta pietiekami sīki, lai rezultātus var
izvērtēt kritiski, neatkarīgi no autora interpretācijas.
Rezultātus skaidrojot, tos var papildināt ar ilustrācijām;
21.5. rezultātu statistisku
analīzi, ja tādu pieprasa testa programma, un datu
variablumu;
21.6. objektīvu iztirzājumu par
iegūtajiem datiem, kas ļauj secināt par vielas:
21.6.1. drošumu,
21.6.2 drošuma robežlielumu
kontroles dzīvniekā un mērķa dzīvniekā, 21.6.3. iespējamām
blakusparādībām,
21.6.4. lietošanas jomu,
21.6.5. aktīvās devas
lielumiem,
21.6.6. jebkādu varbūtēju
nesavietojamību;
21.7. sīki izstrādātu aprakstu un
sīku iztirzājumu par pētījuma rezultātiem attiecībā uz
atliekvielu drošumu pārtikā un to saistību ar atliekvielu
izraisīto iespējamo apdraudējumu cilvēkiem. Šis iztirzājums
atbilst ierosinājumiem ar mērķi nodrošināt, lai samazinātu
jebkādu apdraudējumu cilvēkam, izmantojot starptautiski atzītus
novērtēšanas kritērijus (piemēram, nenovēro ietekmi dzīvniekos,
priekšlikumi attiecībā uz drošības faktora izvēli un pieļaujamo
diennakts devu (PDD));
21.8. sīku iztirzājumu par jebkādu
risku personām, kas gatavo zāles vai lieto tās dzīvniekiem.
Iztirzājumam pievieno priekšlikumus par lietderīgajiem pasākumiem
šādu risku mazināšanā;
21.9. sīku iztirzājumu par jebkādu
risku videi, ko var izraisīt zāļu lietošana piedāvātajos
lietošanas nosacījumos, tai pievieno attiecīgus priekšlikumus
šādu risku samazināšanai;
21.10. vajadzīgās ziņas, lai
klīnicists pēc iespējas pilnīgāk iepazītos ar piedāvāto zāļu
lietderību. Iztirzājumam pievieno ierosinājumus attiecībā uz
blakusparādībām un iespējamu terapiju akūtu toksisku reakciju
gadījumā dzīvniekiem, kuriem lietotas zāles;
21.11. eksperta ziņojumu ar
atzinumu, kurā sniedz sīki izklāstītu kritisku analīzi attiecībā
uz iepriekš minētajām ziņām atbilstoši zinātnes attīstības
līmenim iesnieguma iesniegšanas brīdī, kam pievienots sīki
izstrādāts kopsavilkums par attiecīgo drošuma testu rezultātiem
un precīzas bibliogrāfiskas atsauces.
22. Atliekvielu testēšana:
22.1. šajos noteikumos
“atliekvielas” ir jebkādas aktīvās vielas vai to metabolīti, kas
paliek gaļā vai citos pārtikas produktos, ko iegūst no dzīvnieka,
kam lietotas zāles;
22.2. atliekvielu izpētes mērķis
ir noteikt:
22.2.1. vai atliekvielas
saglabājas pārtikas produktos, kas iegūti no dzīvniekiem, kam
lietotas zāles un, ja saglabājas, tad– kādos apstākļos un kādā
apmērā,
22.2.2. noteikt zāļu izdalīšanās
laiku, kas jāievēro, lai novērstu cilvēku veselības apdraudējumu
un/vai sarežģījumus pārtikas produktu rūpnieciskajā
pārstrādē;
22.3. novērtējot atliekvielu
radīto risku, nosaka, vai ir saglabājušās atliekvielas tādos
dzīvniekos, kas ar zālēm apstrādāti ieteiktajos lietošanas
apstākļos, un izpēta šo atliekvielu iedarbību;
22.4. veterinārajām zālēm, ko
paredzēts lietot produktīviem dzīvniekiem, atliekvielu
dokumentācijā norāda:
22.4.1. kādā mērā un cik ilgi
veterināro zāļu vai to metabolītu atliekvielas saglabājas ar
zālēm apstrādātā dzīvnieka audos vai no tā iegūtajos pārtikas
produktos;
22.4.2. izpildāmus zāļu
izdalīšanās laikus, ko iespējams ievērot praktiskas
lauksaimniecības apstākļos, lai novērstu jebkādu risku apstrādātā
dzīvnieka pārtikas produkta patērētāja veselībai vai sarežģījumus
pārtikas produktu rūpnieciskajā pārstrādē,
22.4.3. ka ir pieejamas praktiskās
analīzes metodes, kas piemērotas regulārai lietošanai, lai
apstiprinātu zāļu izdalīšanās laika ievērošanu.
23. Metabolisms un atliekvielu
kinētika:
23.1. farmakokinētika (absorbcija,
sadalījums, biotransformācija, izdalīšanās):
23.1.1. veterināro zāļu
atliekvielu farmakokinētiskos pētījumus veic nolūkā izvērtēt
produkta absorbciju, sadalījumu, biotransformāciju mērķa sugas
dzīvnieka organismā, kā arī izdalīšanos no tā;
23.1.2. gatavo izstrādājumu vai
bioloģiski līdzvērtīgu formu lieto mērķa sugai maksimāli
ieteicamajā devā;
23.1.3. ievērojot lietošanas
veidu, pilnībā apraksta zāļu absorbcijas apmēru. Ja izrādās, ka
lokāli lietojamu zāļu sistēmiskā absorbcija ir nenozīmīga, nav
nepieciešams veikt tālākus atliekvielu pētījumus;
23.1.4. apraksta zāļu sadalījumu
mērķa dzīvniekā. Apskata iespēju zālēm saistīt plazmas
olbaltumvielu vai pāriet pienā vai olās un uzkrāties lipofīlos
savienojumos;
23.1.5. apraksta zāļu izdalīšanās
ceļus no mērķa dzīvnieka organisma. Identificē un raksturo
galvenos metabolītus;
23.2. atliekvielu izzušana:
23.2.1. pētījumus par atliekvielu
izzušanu mērķa dzīvniekā pēc pēdējās zāļu lietošanas reizes veic
nolūkā noteikt zāļu izdalīšanās laiku;
23.2.2. dažādos laikos pēc tam,
kad testa dzīvnieks ir saņēmis pēdējo veterināro zāļu devu,
nosaka pastāvošos atliekvielu daudzumus, izmantojot attiecīgas
fizikālās, ķīmiskās vai bioloģiskās metodes. Precizē tehniskās
procedūras un lietoto metožu uzticamību un jutīgumu.
24. Regulāra analīzes metode
atliekvielu noteikšanai:
24.1. izmanto analīzes metodes, ko
veic regulāras pārbaudes gaitā un kam ir tāds jutīguma līmenis,
kas ļauj pilnīgi noteikti konstatēt atļauto maksimāli pieļaujamo
zāļu atliekvielu līmeņa pārsniegšanu;
24.2. ierosināto analīzes metodi
sīki apraksta. Tā ir apstiprināta un uzticama lietošanā
parastajos atliekvielu regulārās uzraudzības apstākļos;
24.3. apraksta šādus
raksturlielumus:
24.3.1. specifiskums;
24.3.2. rūpīgums, ietverot
jutību;
24.3.3. precizitāte;
24.3.4. noteikšanas robeža;
24.3.5. daudzumu iedalījuma
robeža;
24.3.6. praktiskums un lietojamība
parastos laboratorijas apstākļos;
24.3.7. jutīgums pret
traucējumiem;
24.4. piedāvāto analīzes metožu
piemērotību izvērtē, atbilstoši zinātnes attīstības līmenim
laikā, kad iesniedz reģistrācijas iesniegumu.
25. Atliekvielu testu
dokumentācijā kā jebkurā zinātniskā darbā iekļauj šādas
ziņas:
25.1. ievadu, kurā definē
aplūkojamo objektu un kuram pievieno noderīgas bibliogrāfiskās
atsauces;
25.2. precīzu zāļu identifikāciju,
ietverot:
25.2.1. sastāvu;
25.2.2. tīrību;
25.2.3. partijas
identifikāciju;
25.2.4. saistību ar gala
produktu;
25.2.5. iezīmēto vielu specifisko
aktivitāti un radioloģisko tīrību;
25.2.6. iezīmēto atomu izvietojumu
molekulā;
25.3. sīki izklāstītu eksperimenta
protokolu, kurā pamato, ja izlaisti kādi no iepriekš minētiem
testiem, izmantoto metožu, aparatūras un materiālu aprakstu,
sīkas ziņas par dzīvnieku sugu, šķirni vai līniju, dzīvnieku
iegūšanu, skaitu, izmitināšanas apstākļiem un barošanas
nosacījumiem;
25.4. iegūtos rezultātus
neatkarīgi no tā, vai tie ir labvēlīgi vai nelabvēlīgi.
Sākotnējos datus apraksta pietiekami sīki, lai varētu rezultātus
izvērtēt kritiski neatkarīgi no autora interpretācijas.
Rezultātiem var pievienot ilustrācijas;
25.5. rezultātu statistisku
analīzi, ja tādu pieprasa testa programma, un datu
variablumu;
25.6. objektīvu iztirzājumu par
iegūtajiem rezultātiem, kam pievienots:
25.6.1. piedāvātais maksimāli
pieļaujamais atliekvielu līmenis aktīvajām vielām zālēs, nosakot
attiecīgās marķieratliekvielas un mērķaudus,
25.6.2. ierosinājums par
nepieciešamajiem zāļu izdalīšanās laikiem, kas nodrošina, ka
pārtikas produktos, kas iegūti no dzīvniekiem, kas apstrādāti ar
zālēm, nav atliekvielu, kas var apdraudēt produktu
patērētāju;
25.7. eksperta ziņojumu ar
atzinumu, kurā sniedz sīki izklāstītu kritisku analīzi attiecībā
uz iepriekš minētajām ziņām atbilstoši zinātnes attīstības
līmenim reģistrācijas iesnieguma iesniegšanas brīdī, kam
pievienots sīki izklāstīts kopsavilkums par atliekvielu testu
rezultātiem un precīzas bibliogrāfiskas atsauces.
IV. Neklīniskie
un klīniskie pētījumi
26. Neklīniskie un klīniskie
pētījumi:
26.1. ziņas un dokumentus, ko
atbilstoši šo noteikumu 13.9. un 13.10.apakšpunktā un 18., 19.,
20. un 21.punktā noteiktajām prasībām pievieno reģistrācijas
iesniegumam, iesniedz saskaņā ar šī pielikuma 27., 28. un
29.punktā izklāstītajām prasībām;
26.2. neklīniskās prasības–
neklīniskie pētījumi ir nepieciešami, lai noteiktu zāļu
farmakoloģisko aktivitāti un toleranci.
27. Farmakoloģija:
27.1. farmakodinamika.
Farmakodinamikas pētījumos ievēro divas atšķirīgas pieejas
jautājumam:
27.1.1. pirmkārt, precīzi apraksta
darbības mehānismu un farmakoloģisko iedarbību, uz ko balstās
ieteiktā lietošana praksē. Rezultātus izsaka kvantitatīvos
kritērijos (piemēram, izmantojot devu un iedarbības līknes, laika
un iedarbības līknes utt.) un pēc iespējas salīdzinājumā ar tādu
vielu, kuras iedarbība ir labi zināma. Ja piesaka kādas aktīvās
vielas stiprāku iedarbību, atspoguļo atšķirību un uzrāda tās
statistisko nozīmīgumu;
27.1.2. otrkārt, pētījumā sniedz
vispārēju aktīvās vielas farmakoloģisku novērtējumu, īpaši
atsaucoties uz iespējamām blakusparādībām. Kopumā pēta galvenās
funkcijas;
27.1.3. pētnieks nosaka lietošanas
veida, formas u.tml. ietekmi attiecībā uz aktīvās vielas
farmakoloģisko iedarbību;
27.1.4. pētījumus pastiprina, ja
ieteiktā deva tuvojas devai, kas var izraisīt
blakusparādības;
27.1.5. eksperimenta metodes, ja
tās nav standarta metodes, apraksta tik sīki, lai tās varētu
reproducēt, un pētnieks veic to apstiprināšanu. Eksperimenta
rezultāti ir skaidri izklāstīti un noteiktu testu gadījumā norāda
to statistisko nozīmīgumu;
27.1.6. ja nav pietiekama
pamatojuma par pretējo, pēta arī visas kvantitatīvās
modifikācijas attiecībā uz reakciju, kas rodas no vielas
atkārtotas lietošanas;
27.1.7. zāļu savienojumus var
lietot farmakoloģiskiem nolūkiem vai klīnisku indikāciju dēļ.
Pirmajā gadījumā farmakodinamikas un/vai farmakokinētikas
pētījumi parāda mijiedarbību, ko var izraisīt pats savienojums
sakarā ar tā klīnisko pielietojumu. Otrajā gadījumā, ja
zinātnisko pamatojumu zāļu savienojumam cenšas iegūt klīnisku
eksperimentu veidā, pētījumos norāda, vai ietekmi, kas gaidāma no
savienojuma, var demonstrēt ar dzīvniekiem. Visbeidzot pārbauda
jebkādu blakusparādību nozīmīgumu. Ja savienojumā ietilpst jauna
aktīvā viela, to iepriekš padziļināti izpēta;
27.2. farmakokinētika:
27.1. parasti klīniskajā kontekstā
noder pamata farmakokinētiskās ziņas attiecībā uz jaunu aktīvo
vielu;
27.2. farmakokinētikas mērķus var
iedalīt divos galvenajos virzienos:
27.2.1. aprakstošā
farmakokinētika, kuras rezultātā novērtē pamatrādītājus, tādus kā
ķermeņa klīrenss, izplatības apjoms, vidējais rezidences laiks
utt.;
27.2.2. šo parametru izmantošana,
lai izpētītu saistību starp devu režīmu, plazmas un audu
koncentrāciju un farmakoloģisku, terapeitisku vai toksisku
iedarbību;
27.3. mērķa sugās parasti veic
farmakokinētiskus pētījumus, lai lietotu zāles ar lielāko
iespējamo iedarbīgumu un drošumu. Tādi pētījumi ir īpaši
noderīgi, lai palīdzētu klīnicistam noteikt devu režīmus
(lietošanas veids un vieta, deva un intervāls starp devām,
ievadīšanas reižu skaits utt.) un pieņemt devas režīmus
atbilstīgi noteiktiem variabliem rādītājiem attiecībā uz
populāciju (piemēram, vecums, slimība). Šādi pētījumi var būt
efektīvāki, tos veicot virknei dzīvnieku, un tie parasti sniedz
vairāk informācijas nekā klasiskie devu titrēšanas pētījumi;
27.4. ja jaunā kombinācijā
lietotas tādas zināmas vielas, kas pētītas saskaņā ar šo
noteikumu prasībām, nav nepieciešams veikt fiksētās kombinācijas
farmakoloģiskus pētījumus, ja iespējams pamatot, ka aktīvo vielu
ievadīšana fiksētas kombinācijas veidā nemaina to
farmakokinētiskās īpašības;
27.5. bioloģiskā pieejamība
(bioekvivalence). Veic attiecīgus bioloģiskās pieejamības
pētījumus, lai noteiktu bioekvivalenci:
27.5.1. ja jaunās formulētās zāles
salīdzina ar jau esošām;
27.5.2. ja jaunu lietošanas veidu
vai paņēmienu salīdzina ar jau pastāvošo;
27.5.3. gadījumos, kas minēti šo
noteikumu 22., 23. un 24.punktā.
28. Tolerance attiecībā uz mērķa
sugas dzīvnieku:
28.1. šā pētījuma mērķis ir visām
dzīvnieku sugām, kurām paredzētas veterinārās zāles, veikt
vietējas un vispārējas tolerances pētījumus, lai noteiktu
panesamo devu, kas ir tik plaša, lai pieļautu pietiekamu drošības
robežlielumu un noteiktu klīniskos nepanesības simptomus,
lietojot ieteikto izmantošanas veidu vai veidus, ciktāl tas ir
iespējams, palielinot terapeitisko devu un/vai lietošanas ilgumu.
Ziņojumā par šiem pētījumiem iekļauj pēc iespējas vairāk precīzu
ziņu par gaidāmajām farmakoloģiskajām iedarbībām un
blakusparādībām. Tās novērtē, ņemot vērā iespēju, ka izmantotie
dzīvnieki var būt ļoti vērtīgi;
28.2. zāles ievada vismaz
ieteiktajā ievadīšanas veidā.
29. Rezistence. Dati par
rezistentu organismu parādīšanos nepieciešami, ja zāles lieto
infekcijas slimību vai parazītu invāzijas profilaksei vai
ārstēšanai dzīvniekiem.
30. Klīniskās prasības:
30.1. klīnisko pētījumu mērķis ir
parādīt vai pamatot veterināro zāļu iedarbību pēc to lietošanas
ieteiktajās devās, noteikt indikācijas un kontrindikācijas
atbilstīgi sugai, vecumam, šķirnei un dzimumam, veterināro zāļu
lietošanas norādījumus, jebkādas blakusparādības, drošumu un
toleranci parastajos lietošanas apstākļos;
30.2. ja nav veikts pamatojums,
klīniskos pētījumus veic ar kontroles dzīvniekiem (kontrolētie
klīniskie pētījumi). Iedarbību salīdzina ar placebo
(konkrētā zāļu formā bioloģiski indiferenta viela, kurai nav
tiešas darbības organismā) vai ar atturēšanās no ārstēšanas
un/vai ar tādu reģistrētu zāļu iedarbību, kuru terapeitiskā
vērtība ir zināma. Ziņo par iegūtajiem rezultātiem;
30.3. norāda metodes, ko izmanto
diagnozes noteikšanai. Rezultātus izklāsta, izmantojot
kvantitatīvus vai pieņemtos klīniskos kritērijus. Lieto un pamato
atbilstīgas statistikas metodes;
30.4. ja veterinārās zāles
galvenokārt paredzēts izmantot par kopējās vērtības uzlabotāju,
īpašu uzmanību pievērš:
30.4.1. dzīvnieka produkcijas
ražībai;
30.4.2. dzīvnieka produkcijas
kvalitātei (organoleptiskās, uztura, higiēniskās un tehnoloģiskās
īpašības);
30.4.3. dzīvnieka uztura
efektivitāte un augšana;
30.4.4. dzīvnieka vispārējais
veselības stāvoklis;
30.5. eksperimentālos datus
apstiprina ar datiem, ko iegūst praktiskajos apstākļos uz
vietas;
30.6. reģistrācijas iesnieguma
iesniedzējs atsevišķos gadījumos attiecībā uz noteiktām
terapeitiskajām indikācijām nespēj sniegt pilnīgus datus par
terapeitisko iedarbību šādu iemeslu dēļ:
30.6.1. indikācijas, kam minētās
zāles paredzētas, sastopamas tik reti, ka no reģistrācijas
iesnieguma iesniedzēja nevar gaidīt pilnīgus pierādījumus;
30.6.2. pastāvošajā zinātnes
attīstības posmā nav iespējams sniegt pilnīgu informāciju;
30.7. Zāļu valsts aģentūra šī
pielikuma 30.6.apakšpunktā minētajā gadījumā veterināro zāļu
reģistrācijas apliecību piešķir ar šādiem nosacījumiem:
30.7.1. minētās zāles izsniedzamas
vienīgi pret veterinārārsta recepti un noteiktos gadījumos to var
ievadīt vienīgi stingrā veterinārārsta uzraudzībā;
30.7.2. lietošanas pamācība un
jebkāda cita informācija vērš veterinārārsta uzmanība uz to, ka
attiecībā uz atsevišķiem aspektiem pieejamās ziņas, kas ir par
zālēm vēl nav pilnīgas.
31. Pētījumu veikšana:
31.1. visus veterināros klīniskos
pētījumus veic saskaņā ar pilnībā noteiktu sīki izklāstītu
izmēģinājuma protokolu, ko rakstiski aizpilda pirms izmēģinājuma
sākšanas. Ievēro pētījumos iesaistīto dzīvnieku labturības
prasības, šie dzīvnieki ir pakļauti veterinārajai uzraudzībai.
Labturības prasības ievēro, sagatavojot pētījumu protokolu un
veicot pētījumus;
31.2. iepriekš izveido sistēmiskas
rakstiskas procedūras klīnisko pētījumu organizēšanai, veikšanai,
datu vākšanai, dokumentēšanai un apstiprināšanai;
31.3. pirms pētījuma uzsākšanas
saņem pētījumā izmantojamo dzīvnieku īpašnieka piekrišanu un to
dokumentē. Dzīvnieka īpašnieku rakstiski informē par to, kādas
sekas pēc piedalīšanās pētījumā rodas attiecībā uz apstrādāto
dzīvnieku likvidāciju vai pārtikas produktu ieguvi no
apstrādātajiem dzīvniekiem. Šā paziņojuma kopiju, ko parakstījis
un datējis dzīvnieka īpašnieks, iekļauj pētījuma
dokumentācijā;
31.4. ja vien pētījumus neveic pēc
aklās metodes, normatīvie akti par veterināro zāļu marķēšanu,
izplatīšanu un kontroli attiecas uz to zāļu formu marķējumu, ko
paredzēts lietot veterinārajos klīniskajos pētījumos. Uz
pētījumos lietoto zāļu marķējuma skaidri un nenodzēšami norāda
frāzi “Lietošanai vienīgi klīniskos veterinārajos pētījumos”.
32. Dokumentos par veterināro zāļu
iedarbīgumu kā jebkurā zinātniskā darbā iekļauj ievadu, kurā
definē aplūkojamo objektu un kuram pievieno izmantojamus
bibliogrāfiskos dokumentus.
33. Neklīniskie un klīniskie
dokumenti ir pietiekami sīki izklāstīti, lai veiktu pamatotu
spriedumu. Ziņo par visiem pētījumiem un izmēģinājumiem
neatkarīgi no tā, vai tie ir reģistrācijas iesnieguma
iesniedzējam labvēlīgi vai nelabvēlīgi.
34. Ziņojumi par neklīniskajiem
novērojumiem pēc iespējas sniedz sīkas ziņas par rezultātiem, kas
iegūti:
34.1. testos, kas demonstrē
farmakoloģiskās darbības;
34.2. testos, kas demonstrē
farmakoloģiskos mehānismus, kuri pakļauti terapeitiskajai
iedarbībai;
34.3. testos, kas demonstrē
galvenos farmakokinētiskos procesus.
35. Ja testēšanas gaitā parādās
kādi neparedzēti rezultāti, par to sniedz sīkas ziņas.
36. Neklīniskajos pētījumos sniedz
arī šādas sīki izklāstītas ziņas:
36.1. kopsavilkumu;
36.2. sīki izstrādātu eksperimenta
protokolu, kurā apraksta izmantotās metodes, aparatūru un
materiālus, dzīvnieku sugu, vecumu, svaru, dzimumu, skaitu un
dzīvnieku šķirni vai līniju, dzīvnieku identifikāciju, zāļu devu,
zāļu lietošanas veidu un grafiku;
36.3. rezultātu statistisko
analīzi;
36.4. objektīvu iztirzājumu par
iegūtajiem rezultātiem, ko izmanto secinājumos par produkta
drošumu un iedarbīgumu.
37. Ja pilnībā vai daļēji izlaiž
kādus datus, to pamato.
38. Ziņojumi par klīniskajiem
novērojumiem:
38.1. visas ziņas iesniedz katrs
pētnieks uz individuālām protokola lapām gadījumā, ja notiek
individuāla apstrāde, un uz kolektīvām protokola lapām, ja notiek
kolektīva apstrāde;
38.2. iesniegtajām ziņām ir šāda
forma:
38.2.1. atbildīgā pētnieka
uzvārds, adrese, amats un kvalifikācija;
38.2.2. apstrādes vieta un datums,
dzīvnieku īpašnieka uzvārds un adrese;
38.2.3. ziņas par pētījuma
protokolu, aprakstot izmantotās metodes, ietverot nejaušās
izvēles un aklo metodi, ziņas par lietošanas veidu, lietošanas
grafiku, devu, pētījumu dzīvnieku identifikāciju, sugu, šķirni
vai līniju, vecumu, svaru, dzimumu, skaitu, fizioloģisko
stāvokli;
38.2.4. audzēšanas un barošanas
metode, aprakstot barības sastāvu un veidu, kā arī jebkādu barībā
esošo piedevu daudzumu;
38.2.5. slimības vēsture (pēc
iespējas pilnīgāka), starplaikā radušos saslimšanu gadījumi un
slimības gaita;
38.2.6. diagnoze un līdzekļi, ar
ko parasti diagnozi nosaka;
38.2.7. slimības simptomi un
smaguma pakāpe pēc iespējas saskaņā ar pieņemtajiem
kritērijiem;
38.2.8. pētījumā izmantotā
klīniskās izpētes formulējuma precīza identifikācija;
38.2.9. zāļu devas, ievadīšanas
veids, paņēmiens un biežums, kā arī piesardzības pasākumi zāļu
lietošanas laikā (injekciju laikā utt.), ja tādi bijuši;
38.2.10. apstrādes un tai sekojošā
novērošanas laikposma ilgums;
38.2.11. visas ziņas par
veterinārajām zālēm (izņemot tās, ko pēta), kas var būt lietotas
pārbaudes laikā vai pirms pētāmām zālēm, vai vienlaikus ar tām,
un pēdējā minētajā gadījumā– sīki izklāstītas ziņas par novēroto
mijiedarbību;
38.2.12. klīnisko pētījumu
rezultāti (ietverot nelabvēlīgus vai negatīvus rezultātus), pilnā
apmērā nosakot klīniskos novērojumus un darbības objektīvo testu
rezultātus (laboratoriskie izmeklējumi, fizioloģiskie testi), kas
nepieciešami, lai novērtētu iesniegumu. Norāda izmantotās metodes
un izskaidro nozīmīgumu jebkādām variācijām rezultātos (piemēram,
metodes variēšanās, variēšanās indivīdu starpā vai zāļu
iedarbībā). Farmakodinamisko efektu norādīšana attiecībā uz
dzīvniekiem nav pietiekama, lai pamatotu atzinumu par jebkādu
terapeitisko iedarbību;
38.2.13. visas ziņas par jebkādu
netīšu ietekmi neatkarīgi no tā, vai tā ir kaitīga vai nav, un
par jebkuriem tā rezultātā veiktiem pasākumiem. Pēc iespējas
izpēta cēloņu un seku sakarību;
38.2.14. ietekme uz dzīvnieku
kopējo vērtību (piemēram, dējību, pienīgumu un reproduktīvajām
funkcijām);
38.2.15. ietekme uz to pārtikas
produktu kvalitāti, kuri iegūti no apstrādātajiem dzīvniekiem, jo
īpaši tādām zālēm, ko paredzēts lietot kā kopējās vērtības
paaugstinātājus;
38.2.16. atzinums par katru
atsevišķo gadījumu, vai, kolektīvas apstrādes gadījumā– uz katru
kolektīvo gadījumu;
38.3. ja nepiemēro vienu vai
vairākas prasības, kas noteiktas no šī pielikuma 38.2.1. līdz
38.2.16.apakšpunktam, to pamato;
38.4. reģistrācijas apliecības
turētājs veic nepieciešamos pasākumus, lai nodrošinātu oriģinālo
dokumentu, kas veido iesniegto datu pamatu, glabāšanu ne mazāk kā
piecus gadus pēc tam, kad veterinārās zāles vairs nav
atļautas.
39. Kopsavilkums un atzinumi par
klīniskajiem novērojumiem. Par katru klīnisko pētījumu klīniskos
novērojumus apkopo pētījumu un to rezultātu sinopsē, jo īpaši
norādot:
39.1. pārbaužu skaitu, to
dzīvnieku skaitu, kas apstrādāti individuāli vai kolektīvi,
sagatavojot dokumentu pa sugām, šķirnēm vai līnijām, pēc vecuma
un dzimuma;
39.2. dzīvnieku skaitu, kas pirms
laika izņemti no pētījumiem, un šādas izņemšanas iemeslus;
39.3. kontroles dzīvniekiem– vai
tie ir:
39.3.1. saņēmuši apstrādi;
39.3.2. saņēmuši placebo;
39.3.3. saņēmuši citas reģistrētas
zāles ar zināmu iedarbību;
39.3.4. saņēmuši pētāmo aktīvo
vielu citā formējumā vai citā lietošanas veidā;
39.4. novēroto blakusparādību
biežumu;
39.5. novērojumus par ietekmi uz
dzīvnieku kopējo vērtību (piemēram, dējību, pienīgumu,
reproduktīvajām funkcijām un pārtikas produktu kvalitāti);
39.6. ziņas par dzīvniekiem, kam
var būt paaugstināts risks, ņemot vērā to vecumu, audzēšanas vai
barošanas veidu, vai nolūku, kam dzīvnieki paredzēti, vai
dzīvnieki, par kuru fizioloģisko vai patoloģisko stāvokli lemj
īpaši;
39.7. rezultātu statistisku
novērtējumu, ja tādu pieprasa testa programma;
39.8. nobeigumā pētnieks izdara
vispārīgus secinājumus no eksperimentālajiem pierādījumiem,
izsakot savu spriedumu par zāļu nekaitīgumu piedāvātajos
lietošanas apstākļos, tā terapeitisko iedarbību un jebkādu
noderīgu informāciju saistībā ar indikācijām un kontrindikācijām,
devām, vidējo apstrādes ilgumu un novērotajām mijiedarbībām ar
citām zālēm vai barības piedevām, kā arī īpašos piesardzības
pasākumus, ko ievēro zāļu lietošanas laikā, un pārdozēšanas
klīniskos simptomus;
39.9. fiksētu kombinēto zāļu
gadījumā pētnieks izdara secinājumus attiecībā uz zāļu drošumu un
iedarbīgumu, to salīdzinot ar tajā iesaistīto aktīvo vielu
atsevišķu lietošanu.
40. Eksperta ziņojums ar atzinumu.
Eksperta ziņojumā ar atzinumu sniedz sīki izklāstītu visas pirms
klīniskās un klīniskās dokumentācijas kritisku analīzi atbilstoši
zinātnes attīstības līmenim reģistrācijas pieteikuma iesniegšanas
brīdī. Tam pievienots sīki izstrādāts kopsavilkums par testu un
pētījumu rezultātiem un precīzas bibliogrāfiskas atsauces.
Zemkopības ministra vietā –
iekšlietu ministrs Dz.Jaundžeikars
Zemkopības
ministrijas iesniegtajā redakcijā
3.pielikums
Ministru kabineta
2006.gada 18.jūlija noteikumiem
Nr.600
Prasības
imunoloģiskajām veterinārajām zālēm
I.
Dokumentācijas kopsavilkums
1. Administratīvie dati:
1.1. imunoloģiskās veterinārās
zāles, uz kurām attiecas reģistrācijas iesniegums, identificē ar
zāļu nosaukumu un aktīvās vielas nosaukumu, norādot zāļu
stiprumu, zāļu formu, ievadīšanas veidu un lietošanas veidu, kā
arī gala realizācijas noformējuma aprakstu;
1.2. uzrāda reģistrācijas
iesnieguma iesniedzēja nosaukumu un adresi, ražotāja nosaukumu un
adresi un objektus, kas iesaistīti dažādajos ražošanas posmos,
ietverot galaprodukta ražotāju un aktīvās vielas ražotāju, kā
arī– attiecīgā gadījumā– importētāja nosaukumu un adresi;
1.3. reģistrācijas iesnieguma
iesniedzējs precīzi norāda dokumentu skaitu un nosaukumu, ko
pievieno reģistrācijas iesniegumam, un informāciju par
iesniegtajiem paraugiem, ja tādi ir;
1.4. administratīvajiem datiem
reģistrācijas iesniegumā pievieno;
1.4.1. dokumentu kopijas, kas
pierāda ražotāja tiesības ražot imunoloģiskas veterinārās zāles
atbilstoši normatīvajiem aktiem par kārtību, kādā izsniedz,
aptur, pārreģistrē un anulē speciālās atļaujas (licences)
veterinārfarmaceitiskai darbībai,
1.4.2. organismu sarakstu, ar ko
rīkojas ražošanas objektā;
1.5. reģistrācijas iesnieguma
iesniedzējs iesniedz to valstu sarakstu, kurās viņam piešķirta
atļauja, kopijas no to veterināro zāļu aprakstiem saskaņā ar šo
noteikumu 35.punkta prasībām, kurus apstiprinājušas dalībvalstis,
un to dalībvalstu sarakstu, kurās iesniegts reģistrācijas
iesniegums.
2. Zāļu īss raksturojums, kad
reģistrācijas iesnieguma iesniedzējs:
2.1. piedāvā zāļu aprakstu saskaņā
ar šo noteikumu 35.punkta prasībām;
2.2. uzrāda vienu vai vairākus
imunoloģisko veterināro zāļu paraugus vai reklāmas maketus un
lietošanas instrukciju, ja tā nepieciešama.
3. Eksperta ziņojumi:
3.1. saskaņā ar šo noteikumu
35.punkta prasībām iesniedz eksperta ziņojumus par visiem
dokumentācijas aspektiem;
3.2. katru eksperta ziņojumu veido
saskaņā ar šo noteikumu prasībām veikts kritisks testu un
pētījumu izvērtējums, kurā iekļauj datus, kuri attiecas uz
izvērtējumu. Eksperts sniedz savu atzinumu par to, vai ir
pietiekamas garantijas attiecīgo zāļu kvalitātei, drošumam un
iedarbīgumam. Faktu kopsavilkums bez atzinuma ir
nepietiekams;
3.3. visus būtiskos datus apkopo
eksperta ziņojuma pielikumā, ja iespējams, tabulas vai grafiskā
veidā. Eksperta ziņojumā un kopsavilkumos ir precīzas
savstarpējās norādes uz ziņām, kas iekļautas pamatdokumentos;
3.4. katru eksperta ziņojumu
sagatavo atbilstoši kvalificēta persona, kurai ir atbilstoša
pieredze. Eksperta ziņojumu paraksta un datē eksperts, un
ziņojumam pievieno īsas ziņas par eksperta izglītību un
profesionālo pieredzi. Norāda eksperta un pieteikuma iesniedzēja
profesionālās attiecības.
II. Imunoloģisku
veterināro zāļu analītiskie (fizikāli ķīmiskie, bioloģiskie vai
mikrobioloģiskie) testi
4. Pielietotās testēšanas
procedūras atbilst tā brīža zinātnes progresam un ir
apstiprinātas procedūras. Norāda apstiprināšanas pētījumu
rezultātus.
5. Testēšanas procedūru
(procedūras) sīki apraksta, lai šīs procedūras var atkārtot
kontroles testos, ko veic pēc Zāļu valsts aģentūras pieprasījuma.
Sīki apraksta izmantojamās speciālās iekārtas un aprīkojumu, pēc
iespējas pievienojot shēmu. Ja nepieciešams, laboratorijas
reaģentu formulu aprakstus papildina ar pagatavošanas metodes
aprakstu. Ja testēšanas procedūras iekļautas Eiropas Farmakopejā
vai dalībvalsts farmakopejā, testēšanas procedūru aprakstu var
aizstāt ar sīki izklāstītu atsauci uz Eiropas Farmakopeju vai
dalībvalsts farmakopeju.
6. Reģistrācijas iesniegumam
pievienotās šo noteikumu 13.3.apakšpunktā noteiktās ziņas un
dokumentus par veterināro zāļu sastāvā ietilpstošo aktīvo vielu
un sastāvdaļu kvalitatīvo un kvantitatīvo sastāvu iesniedz
saskaņā ar šādiem nosacījumiem:
6.1. imunoloģisko veterināro zāļu
visu komponentu kvalitatīvo ziņu jēdziens ietver to, ka ir
nosaukta un aprakstīta:
6.1.1. aktīvā viela (vielas);
6.1.2. palīgvielu komponenti;
6.1.3. palīgvielu komponenti
neatkarīgi no to veida un daudzuma. Palīgvielu komponenti ietver
konservantus, stabilizatorus, emulgatorus, krāsvielas, garšas un
aromātiskās vielas, kontrastvielas u.tml.;
6.1.4. dzīvniekiem lietoto zāļu
formu komponenti;
6.2. informācija par trauku un tā
noslēgšanas sistēmu vienlaikus ar datiem par ierīcēm imunoloģisku
veterināro zāļu lietošanai un ievadīšanai, ko piegādā komplektā
ar šīm zālēm;
6.3. “parastā terminoloģija”, ko
izmanto, lai aprakstītu imunoloģisku veterināro zāļu komponentus
neatkarīgi no šo noteikumu 13.3.apakšpunkta pārējo nosacījumu
piemērošanas, nozīmē:
6.3.1. vielām, kas ierakstītas
Eiropas Farmakopejā vai, ja tās nav, dalībvalsts farmakopejā–
attiecīgās farmakopejas monogrāfijā noteikto vielas
pamatnosaukumu, kas kļūst obligāts visām šīm vielām, ar atsauci
uz attiecīgo farmakopejas monogrāfiju;
6.3.2. pārējām vielām – Pasaules
Veselības organizācijas ieteikto starptautisko nepatentēto
nosaukumu, kam var būt pievienots kāds cits nepatentēts
nosaukums, vai, ja tāda nav, precīzs zinātniskais nosaukums.
Vielas, kam nav starptautiska nepatentēta nosaukuma vai precīza
zinātniskā nosaukuma, apraksta, nosakot, kā un no kā tās
pagatavotas, attiecīgi papildinot ar citām svarīgām ziņām;
6.3.3. attiecībā uz krāsvielu
apzīmējums ar “E” kodu, kas tām noteikts atbilstoši šo noteikumu
4.pielikuma prasībām;
6.4. kvantitatīvās ziņas:
6.4.1. lai sniegtu imunoloģisku
veterināro zāļu aktīvo vielu kvantitatīvās ziņas, pēc iespējas
jānorāda:
6.4.1.1. organismu skaits,
specifisko olbaltumvielu saturs, masa, starptautisko vienību (SV)
vai bioloģiskās aktivitātes vienību skaits devas vienībā vai
tilpumā,
6.4.1.2. palīgvielai un
palīgvielas komponentiem masa un tilpums (katrai no tām)
atbilstoši šī pielikuma 8.punktā noteiktajām ziņām;
6.4.2. ja ir noteikta
starptautiskā bioloģiskās aktivitātes vienība, lieto to,
6.4.3. bioloģiskās aktivitātes
vienība, par kuru nav publicētu datu, izsaka sniedzot
nepārprotamu informāciju par sastāvdaļu aktivitāti, piemēram,
nosakot imunoloģisko iedarbību, uz ko balstās devas noteikšanas
metode.
7. Farmaceitiskā izstrādnē sniedz
skaidrojumu par veterināro imunoloģisko zāļu sastāvu, sastāvdaļām
un iepakojumu, pievienojot zinātniskos datus par farmaceitisko
izstrādni. Nosaka devas palielinājumu, to pamatojot. Pierāda
jebkuras konservantu sistēmas efektivitāti.
8. Gatavās produkcijas ražošanas
metodes aprakstu sagatavo atbilstoši šādiem nosacījumiem:
8.1. ražošanas metodes apraksts,
ko atbilstoši šo noteikumu 13.4.apakšpunktam pievieno
reģistrācijas iesniegumam, sniedz pietiekamu pārskatu par
ražošanas operāciju raksturu;
8.2. ražošanas metodes aprakstā
iekļauj vismaz šādas ziņas:
8.2.1. ataino dažādus ražošanas
posmus (ietverot attīrīšanas procedūras), lai var veikt
novērtējumu par:
8.2.1.1. ražošanas procedūras
atkārtojamību,
8.2.1.2. blakusparādību risku
gatavajā produkcijā kā mikrobioloģiskā piesārņojuma risku,
8.2.2. nepārtrauktās ražošanas
gadījumā– pilnīgas ziņas par piesardzības pasākumiem, ko veic,
lai nodrošinātu gatavās produkcijas visu sēriju homogēnumu un
viendabību;
8.2.3. nosauc vielas, kas
neatjaunojas ražošanas gaitā;
8.2.4. sniedz sīki izklāstītu
informāciju par zāļu samaisīšanu ar kvantitatīvām sīkām ziņām par
visām izmantotajām vielām;
8.2.5. norāda ražošanas posmu,
kurā ņem paraugu kontrolei ražošanas gaitā.
9. Izejmateriālu ražošana un
kontrole:
9.1. šī punkta izpratnē
“izejmateriāli” nozīmē sastāvdaļas, ko lieto imunoloģisku
veterināro zāļu ražošanā. Kultivēšanas barotnes, ko izmanto
aktīvās vielas ražošanā, uzskata par vienotu izejmateriālu;
9.2. ja cita persona, nevis
reģistrācijas iesnieguma iesniedzējs, ražo aktīvo vielu, kas nav
aprakstīta Eiropas Farmakopejā vai dalībvalsts farmakopejā, vai
aktīvo vielu, kas aprakstīta Eiropas Farmakopejā vai dalībvalsts
farmakopejā, bet tās pagatavošanas metode var atstāt
piemaisījumus, kuri nav minēti farmakopejas monogrāfijā un kuru
atbilstīgai kvalitātes kontrolei monogrāfija nav piemērota,
reģistrācijas iesnieguma iesniedzējs var pieprasīt aktīvās vielas
ražotājam iesniegt Zāļu valsts aģentūrā sīki izstrādātu aprakstu
par ražošanas metodi, kvalitātes kontroli ražošanas gaitā un
procesa apstiprināšanu;
9.3. šī pielikuma 9.2.apakšpunktā
minētajā gadījumā aktīvās vielas ražotājs sniedz reģistrācijas
iesnieguma iesniedzējam ziņas, kas tam nepieciešamas, lai
uzņemtos atbildību par veterinārajām zālēm. Aktīvās vielas
ražotājs rakstiski apstiprina reģistrācijas iesnieguma
iesniedzējam, ka ražotājs nodrošina visu sēriju atbilstību
paraugam un bez reģistrācijas iesnieguma iesniedzēja ziņas
nemaina ražošanas procesu vai specifikācijas. Zāļu valsts
aģentūrā reģistrācijas iesnieguma iesniedzējs vai ražotājs
iesniedz dokumentus un ziņas, ko pievieno reģistrācijas
iesnieguma dokumentācijai, par šādām izmaiņām;
9.4. ziņās un dokumentos, ko
pievieno reģistrācijas iesniegumam atbilstoši šo noteikumu 13.9.
un 13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām, iekļauj izmantoto sastāvdaļu kvalitātes kontroles testu
rezultātus un iesniedz saskaņā ar šeit uzskaitītiem
nosacījumiem:
9.4.1. par izejmateriāliem, kas
uzskaitīti farmakopejās:
9.4.1.1. Eiropas Farmakopejas
monogrāfijas attiecas uz visām tajā sastopamajām vielām;
9.4.1.2. attiecībā uz citām vielām
katra dalībvalsts var pieprasīt, lai attiecībā uz tās teritorijā
ražotajām zālēm tiktu ievērota tās nacionālā farmakopeja;
9.4.1.3. sastāvdaļas, kas atbilst
Eiropas Farmakopejas prasībām vai dalībvalsts farmakopejas
prasībām, uzskata par pietiekamā mērā atbilstošām šo noteikumu
13.9.apakšpunkta prasībām. Šajā gadījumā analītisko metožu
aprakstu var aizstāt ar precīzu atsauci uz minēto
farmakopeju;
9.4.1.4. var atļaut atsauces uz
trešo valstu farmakopejām, ja attiecīgā viela nav aprakstīta
Eiropas Farmakopejā. Šādā gadījumā iesniedz trešās valsts
farmakopejas monogrāfijas, kam nepieciešamības gadījumā pievieno
tulkojumu, par ko ir atbildīgs reģistrācijas iesnieguma
iesniedzējs;
9.4.2. krāsvielas atbilst šo
noteikumu 4.pielikuma prasībām;
9.4.3. regulārie testi, ko veic
katrai izejmateriālu sērijai, ir tādi, kā deklarēts reģistrācijas
iesniegumā. Ja izmanto testus, kas nav minēti farmakopejā, tad
sniedz pierādījumus, ka izejmateriāli atbilst farmakopejā
noteiktajām kvalitātes prasībām;
9.4.4. ja specifikācija vai citi
noteikumi, kas ierakstīti Eiropas Farmakopejā vai dalībvalsts
farmakopejā, var nebūt pietiekami, lai nodrošinātu vielas
kvalitāti, kompetentās iestādes var pieprasīt no reģistrācijas
iesnieguma iesniedzēja atbilstošas specifikācijas;
9.4.5. Zāļu valsts aģentūra
informē iestādes, kuras atbildīgas par attiecīgo farmakopeju.
Reģistrācijas iesnieguma iesniedzējs sniedz šo farmakopeju
pārzinošajām iestādēm sīkas ziņas par varbūtējo nepilnību un par
papildus izmantotajām specifikācijām;
9.4.6. ja izejmateriāls nav
aprakstīts Eiropas Farmakopejā vai dalībvalsts farmakopejā, Zāļu
valsts aģentūra var akceptēt atbilstību monogrāfijai kādas trešās
valsts farmakopejā. Šādos gadījumos reģistrācijas iesnieguma
iesniedzējs nodrošina monogrāfijas kopiju, kam, ja nepieciešams,
pievienots monogrāfijā noteikto testēšanas procedūru
apstiprinājums un tulkojums. Tiek parādīts, ka monogrāfija spēj
pietiekami kontrolēt aktīvo sastāvdaļu kvalitāti.
10. Izejmateriāli, kas nav
uzskaitīti farmakopejā:
10.1. bioloģiskas izcelsmes
izejmateriāli:
10.1.1. aprakstu sniedz
monogrāfijas veidā;
10.1.2. vakcīnas ražošanu pēc
iespējas balsta uz sējmateriāla sistēmu un uz esošajām šūnu
bankām. Ražojot imunoloģiskās veterinārās zāles, kas sastāv no
seruma, norāda ieguvē izmantotā dzīvnieka vispārējo veselības
stāvokli un imunoloģisko stāvokli. Izmanto noteiktus
izejmateriālu kopumus;
10.1.3. apraksta un dokumentē
izejmateriālu izcelsmi un vēsturi. Par gēnu inženierijas
materiāliem šajā informācijā iekļauj ziņas: izejas šūnu vai celmu
apraksts, vektora uzbūves apraksts (nosaukums, izcelsme,
replikona funkcija, aktivatora paātrinātājs un citi regulējošie
elementi), efektīvi iekļauta DNS vai RNS sekvences kontrole,
plazmīdu vektora oligonukleotīdās sekvences šūnās, kotransfekcijā
izmantotās plazmīdas, pievienotie vai izņemtie gēni, gatavās
struktūras un izdalīto gēnu bioloģiskās īpašības, pavairojumu
skaits un ģenētiskā stabilitāte;
10.1.4. kultūras materiālu,
ietverot šūnu bankas un izejmateriāla serumu antiseruma
pagatavošanai, testē, lai noteiktu identitāti un papildu
aģentus;
10.1.5. sniedz ziņas par
bioloģiskas izcelsmes vielām, ko lieto katrā ražošanas procesa
posmā. Šīs ziņas ietver:
10.1.5.1. sīkas ziņas par
materiālo resursu avotu;
10.1.5.2. sīkas ziņas par
pārstrādi, attīrīšanu un inaktivāciju, pievienojot šo procesu
apstiprināšanas datus un kontroli ražošanas gaitā;
10.1.5.3. sīkas ziņas par testiem,
ko veic katrai vielas sērijai attiecībā uz piesārņojumu;
10.1.6. ja tiek atklāta papildu
aģentu klātbūtne vai pastāv aizdomas par tādu klātbūtni,
attiecīgo materiālu novirza atkritumos vai lieto ārkārtējos
izņēmuma gadījumos, ja produkta tālāka apstrāde nodrošina
papildus aģenta izzušanu un (vai) inaktivāciju. Pierāda šādu
papildu aģentu izzušanu un (vai) inaktivāciju;
10.1.7. ja izmanto šūnu bankas,
atspoguļo šūnu raksturlielumu nemainīgumu līdz pat ražošanā
lietotās pasāžas augstākajam līmenim;
10.1.8. par dzīvām novājinātām
vakcīnām sniedz pierādījums par uzsējuma novājinājuma īpašību
stabilitāti;
10.1.9. ja nepieciešams, iesniedz
bioloģiskā izejmateriāla vai testēšanas procedūrā izmantoto
reaģentu paraugus, lai Zāļu valsts aģentūra var vienoties par
pārbaudes testu veikšanu;
10.2. izejmateriāliem, kuru
izcelsme nav bioloģiska, aprakstu sniedz monogrāfijas veidā ar
šādām pozīcijām:
10.2.1. izejmateriāla nosaukumam,
kas atbilst šī pielikuma 9.1.apakšpunkta prasībām, pievieno
komerciālos vai zinātniskos sinonīmus;
10.2.2. izejmateriāla apraksts
izklāstīts līdzīgā veidā, kā to veic Eiropas Farmakopejas
apraksta punktā;
10.2.3. izejmateriāla
funkcija;
10.2.4. identifikācijas
metodes;
10.2.5. tīrības pakāpe aprakstīta
saistībā ar kopējo daudzumu, ko veido prognozētie piemaisījumi,
īpaši tādi, kam var būt kaitīgas sekas, un tie, kas var negatīvi
ietekmēt zāļu stabilitāti vai izkropļot analīzes rezultātus,
ņemot vērā vielu salikumu, uz kuru attiecas pieteikums. Sagatavo
īsu aprakstu par testiem, ko veic, lai noteiktu katras
izejmateriāla sērijas tīrību;
10.2.6. norāda īpašos piesardzības
pasākumus, kas var būt nepieciešami izejmateriāla uzglabāšanai,
un vajadzības gadījumā maksimāli pieļaujamo uzglabāšanas
laikposmu.
11. Īpaši pasākumi, lai novērstu
dzīvnieku sūkļveida encefalopātijas pārnešanu– reģistrācijas
iesnieguma iesniedzējs uzrāda, ka veterinārās zāles ir ražotas
saskaņā ar Komisijas Norādījumiem par riska samazināšanu līdz
minimumam attiecībā uz iespēju veterinārām zālēm pārnēsāt
dzīvnieku sūkļveida encefalopātijas izraisītājus, un norādījumu
jaunākajām redakcijām, kuras Eiropas Komisija publicējusi Eiropas
Kopienas Zāļu reglamentācijas noteikumu 7.sējumā.
12. Ražošanas starpposmos veiktie
kontroles testi:
12.1. ziņās un dokumentos, ko
pievieno reģistrācijas iesniegumam, atbilstoši šo noteikumu 13.9.
un 13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām, iekļauj ziņas par kontroles testiem, ko veic
produkcijai kādā ražošanas starpposmā, lai nodrošinātu ražošanas
procesa un gala produkta atbilstību;
12.2. inaktivētām vai
detoksificētām vakcīnām inaktivāciju un detoksikāciju testē katra
ražošanas cikla laikā tūlīt pēc inaktivācijas vai detoksikācijas
procedūras pabeigšanas.
13. Gatavās produkcijas
kontrole:
13.1. ziņās un dokumentos, ko
pievieno reģistrācijas iesniegumam atbilstoši šo noteikumu 13.9.
un 13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām, iekļauj ziņas par kontroli, ko veic gatavajai
produkcijai. Ja, pastāvot atbilstīgām monogrāfijām, lieto citas
testēšanas procedūras un robežlielumu, nevis tos, kas ierakstīti
Eiropas Farmakopejas monogrāfijās, tad sniedz pierādījumu, ka
gatavais izstrādājums tā testēšanas gadījumā saskaņā ar šo
monogrāfiju atbilst šādas farmakopejas kvalitātes prasībām par
konkrēto farmaceitisko formu. Reģistrācijas iesniegumā uzskaita
tos testus, ko veic katras gatavās produkcijas sērijas
raksturīgiem paraugiem. Nosaka to testu biežumu, kurus neveic
katrai sērijai. Norāda izlaides robežas;
13.2. gatavā izstrādājuma
vispārīgās īpašības:
13.2.1. gatavā izstrādājuma
testēšanā iekļauj noteiktus testus par izstrādājuma vispārīgām
īpašībām, pat ja tie veikti ražošanas procesa gaitā;
13.2.2. šie testi attiecas uz
vidējā svara un maksimālās novirzes kontroli, uz mehāniskiem,
fizikāliem, ķīmiskiem vai mikrobioloģiskiem testiem, fiziskajām
īpašībām, tādām kā blīvums, pH, atstarošanas indekss utt.
Reģistrācijas iesnieguma iesniedzējs katrā atsevišķajā gadījumā
nosaka specifikācijas ar pienācīgām pielaides robežām katrai no
nosauktajām īpašībām;
13.3. aktīvās vielas
identifikācija un novērtējums:
13.3.1. visiem testiem gatavās
produkcijas analīzes metodes aprakstu izklāsta sīki, lai to var
atkārtot;
13.3.2. aktīvās vielas bioloģiskās
aktivitātes novērtējumu produkcijas sērijas raksturīgā paraugā
vai arī virknē dozējuma vienību, ko analizē individuāli;
13.3.3. vajadzības gadījumā veic
arī īpašu identifikācijas testu;
13.3.4. izņēmuma gadījumos ar
īpaši sarežģītiem maisījumiem, ja ļoti daudzskaitlīgu vai ļoti
mazos daudzumos pārstāvētu aktīvo vielu novērtēšanai vajadzīgs
sarežģīts pētījums, ko grūti veikt katrai produkcijas sērijai,
gatavajā produkcijā var neveikt vienas vai vairāku aktīvo vielu
novērtēšanu, ar skaidru nosacījumu, ka šādus novērtējumus veic
pēc iespējas vēlākajos ražošanas procesa starpposmos. Šādu atkāpi
prasībās neattiecina uz šo vielu raksturojumu. Šo vienkāršoto
metodi papildina ar kvantitatīvā izvērtējuma metodi, kas ļauj
Zāļu valsts aģentūrai pārliecināties par tirgū laistu
imunoloģisku veterināro zāļu atbilstību formulai.
14. Palīgvielu identifikācija un
novērtējums– ciktāl ir pieejamas testēšanas procedūras, pārbauda
papildvielas un tās sastāvdaļu daudzumu un veidu gatavajā
produktā.
15. Palīgvielas sastāvdaļu
identifikācija un novērtējums:
15.1. ciktāl nepieciešams,
palīgvielai veic vismaz identifikācijas testus;
15.2. testēšanas procedūra, ko
piedāvā krāsvielu identificēšanai, ļauj pārliecināties, ka šādas
vielas ir atļautas atbilstoši šo noteikumu 4.pielikuma
prasībām;
15.3. obligāti veic augšējās un
apakšējās robežas testu par konservantiem. Augšējās robežas tests
ir obligāts attiecībā uz jebkādu citu papildvielas sastāvdaļu,
kas var izraisīt blaknes.
16. Drošuma testi– līdztekus tiem
testu rezultātiem, kas iesniegti saskaņā ar šī pielikuma III
daļu, iesniedz sīkas ziņas par drošuma testiem. Šie testi ir
pētījumi par zāļu pārdozēšanu, tos veic ne mazāk kā:
16.1. ar vienu no jutīgākajām
mērķa sugām;
16.2. ieteiktajā ievadīšanas
veidā, kas rada vislielāko risku.
17. Sterilitātes un tīrības tests–
attiecīgi testi, lai pārliecinātos par to, ka nav piesārņojuma ar
papildu aģentiem vai citām vielām, veic atbilstīgi imunoloģisko
veterināro zāļu īpašībām, ražošanas metodei un apstākļiem.
18. Inaktivācija– tests
produkcijai galīgajā iepakojumā, lai apstiprinātu
inaktivāciju.
19. Atlikuma mitrums– katrai
liofilizēta izstrādājuma sērijai pārbauda atlikuma mitrumu.
20. Visu sēriju atbilstība
paraugam– lai nodrošinātu, ka produkta iedarbību iespējams
reproducēt preču sērijām un pierādītu atbilstību specifikācijām,
katrai nedalītai gala izstrādājuma (kravas) sērijai vai gala
produkta sērijai veic potenču testēšanu, kas balstās uz in
vitro vai in vivo metodēm, piemērojot atsauču
materiālus ar attiecīgu ticamības pakāpi, ja tādi ir pieejami.
Ārkārtas apstākļos potencēšanas testus var veikt pēc iespējas
vēlākā ražošanas procesa starpposmā.
21. Stabilitātes testi:
21.1. ziņas un dokumentus, ko
pievieno reģistrācijas iesniegumam atbilstoši šo noteikumu 13.9.
un 13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām, iesniedz saskaņā ar šādām prasībām:
21.1.1. apraksta testus, kas
veikti, lai pārliecinātos par reģistrācijas iesnieguma
iesniedzēja piedāvāto derīguma termiņu. Šie testi vienmēr ir
reālā laika pētījumi. Tos veic ar pietiekamu daudzumu preču
sēriju, kas saražotas saskaņā ar aprakstīto ražošanas procesu, un
ar izstrādājumiem, ko uzglabā gala iepakojumā. Šie testi ietver
bioloģiskās un fizikāli ķīmiskās stabilitātes testus;
21.1.2. atzinumos ir analīžu
rezultāti, kas pamato piedāvāto glabāšanas termiņu visos
piedāvātajos uzglabāšanas apstākļos;
21.1.3. zālēm, ko ievada ar
barību, ja nepieciešams, sniedz informāciju par produkta derīguma
termiņu dažādās piemaisīšanas fāzēs, ja piemaisīšanu veic saskaņā
ar ieteikto lietošanas pamācību;
21.2. ja pirms ievadīšanas
nepieciešams gatavās zāles atjaunot, nepieciešamas sīkas ziņas
par atjaunoto zāļu ieteicamo derīguma termiņu. Iesniedz datus,
kas pamato ieteikto uzglabāšanas termiņu atjaunotam
izstrādājumam.
III. Drošuma
testēšana
22. Drošuma testi parāda iespējamo
risku, ko var izraisīt imunoloģisks veterinārais produkts, to
lietojot dzīvniekiem ieteiktajos lietošanas apstākļos. Iespējamo
risku novērtē attiecībā pret iespējamo ieguvumu, ko var dot
imunoloģisks veterinārais produkts.
23. Ja imunoloģiskas veterinārās
zāles satur dzīvus organismus, jo īpaši tādus, kurus var izplatīt
vakcinētie dzīvnieki, novērtē potenciālo risku nevakcinētiem
dzīvniekiem, kas pieder pie tās pašas vai kādas citas iedarbībai
iespējami pakļautas sugas.
24. Ziņas un dokumentus, ko
pievieno reģistrācijas iesniegumam atbilstoši šo noteikumu 13.9.
un 13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām, iesniedz saskaņā ar šī pielikuma 26.punkta
prasībām.
25. Laboratorijas testus veic
atbilstoši labas laboratoriskās prakses principiem, kas
izklāstīti normatīvajos aktos par prasībām laboratorijas darba
kvalitātei un laboratoriju inspicēšanai.
26. Vispārīgās prasības:
26.1. drošuma testus veic mērķa
sugai;
26.2. izmantojamo zāļu deva ir
daudzums, ko ieteic lietošanai, un tas satur maksimālo titru vai
potenci, par ko ir iesniegts reģistrācijas iesniegums;
26.3. paraugu, ko izmanto drošuma
testēšanai, ņem no sērijas vai sērijām, kas saražota saskaņā ar
ražošanas procesu, kas aprakstīts reģistrācijas iesniegumā.
27. Laboratorijas testi:
27.1. drošums vienas devas
piemērošanas gadījumā:
27.1.1. imunoloģiskās veterinārās
zāles lieto ieteiktajā devā un katrā no ieteiktajiem lietošanas
veidiem visu to sugu un kategoriju dzīvniekiem, kam to paredzēts
izmantot, ietverot dzīvniekus ar mazāko pieļaujamo vecumu zāļu
lietošanai. Dzīvniekus novēro un izmeklē, lai noteiktu sistēmisku
un lokālu reakciju pazīmes. Šajā izpētē ietilpst rūpīgi veikta
pēckaušanas makroskopiska un mikroskopiska apskate injekcijas
vietā. Reģistrē citus objektīvus kritērijus, kā rektālo
temperatūru un kopējās vērtības mērījumus;
27.1.2. dzīvniekus novēro, līdz
vairs nav gaidāma reakcija, bet visos gadījumos novērošanas un
izmeklēšanas laika posms ir ne mazāks kā 14 dienas pēc zāļu
ievadīšanas;
27.2. vienas lietošanas drošums
attiecībā pret pārdozēšanu:
27.2.1. imunoloģisku veterināro
zāļu pārsniegtu devu piemēro katrā no ieteiktajiem ievadīšanas
veidiem dzīvniekiem no visjutīgākās mērķa sugas kategorijas.
Dzīvniekus novēro un izmeklē, lai noteiktu sistēmiskas un lokālas
reakciju pazīmes. Reģistrē citus objektīvus kritērijus, kā
rektālo temperatūru un kopējās vērtības mērījumus;
27.2.2. dzīvniekus novēro un
izmeklē ne mazāk kā 14 dienas pēc zāļu lietošanas;
27.3. drošums vienas devas
atkārtotas piemērošanas gadījumā:
27.3.1. vienas devas atkārtota
lietošana nepieciešama, lai atklātu jebkādas blakusparādības, ko
izraisa šāda lietošana. Testus veic visjutīgākajai mērķa sugas
kategorijai, izmantojot ieteikto ievadīšanas veidu;
27.3.2. dzīvniekus novēro un
apskata ne mazāk kā 14 dienas pēc pēdējās zāļu ievadīšanas, lai
noteiktu sistēmiskas un lokālas reakcijas pazīmes. Reģistrē citus
objektīvus kritērijus, kā rektālo temperatūru un kopējās vērtības
mērījumus.
28. Reproduktīvo funkciju
pārbaude:
28.1. par reproduktīvo funkciju
pārbaudi lemj, ja dati liecina, ka zāļu izejmateriāls var būt
potenciāls riska faktors. Reproduktīvo funkciju vīriešu kārtas
dzīvniekiem un grūsniem un negrūsniem sieviešu kārtas dzīvniekiem
pēta ar ieteikto devu un ar katru no ieteiktajiem ievadīšanas
veidiem. Izpēta kaitīgo ietekmi uz pēcnācējiem, kā arī teratogēno
un abortu izraisošo ietekmi;
28.2. šie pētījumi var veidot šī
pielikuma 27.1.apakšpunktā aprakstītās drošuma izpētes
sastāvdaļu.
29. Imunoloģisko funkciju
pārbaude– ja imunoloģiskām veterinārajām zālēm var būt kaitīga
ietekme uz vakcinētā dzīvnieka vai to pēcnācēju imūnreakciju,
veic attiecīgus testus, pārbaudot imunoloģiskās funkcijas.
30. Īpašas prasības dzīvām
vakcīnām:
30.1. vakcīnas celma izplatīšanās–
izpēta vakcīnas celma izplatīšanos no vakcinētiem uz
nevakcinētiem mērķa dzīvniekiem, izmantojot ieteikto ievadīšanas
veidu, kas visdrīzāk var izraisīt vakcīnas celma izplatīšanos.
Turklāt var būt nepieciešams izpētīt izplatīšanos uz sugām, kas
nav mērķa sugas un kas var būt visjutīgākās attiecībā pret dzīvu
vakcīnas celmu;
30.2. izplatīšanās vakcinētajā
dzīvniekā– pārbauda izkārnījumus, urīnu, pienu, olas, deguna un
citus izdalījumus, lai konstatētu tajos organismus. Iespējams, ka
ir nepieciešams veikt pētījumus par vakcīnas celma izplatīšanos
ķermenī, īpašu uzmanību pievēršot ķermeņa vietām, kas disponētas
organisma replikācijai. Šos pētījumus veic, ja produktīviem
dzīvniekiem lieto dzīvas vakcīnas pret labi zināmām zoonozēm;
30.3. novājināto vakcīnu
virulences reversija– virulences reversiju pēta ar materiālu no
tā pasāžas līmeņa, kurš ir vismazāk novājināts starp celma
kultūru un gala produktu. Sākotnējo vakcināciju veic, izmantojot
ieteikto ievadīšanas veidu, kas visdrīzāk var izraisīt virulences
reversiju. Veic ne mazāk kā piecas sērijas pasāžas mērķa sugas
dzīvniekos. Ja tas tehniski nav iespējams, jo organisma
replikācija ir nepietiekama, veic tik pasāžu, cik iespējams mērķa
sugā. Vajadzības gadījumā starp pasāžām in vivo var veikt
organisma izplatīšanu in vitro. Pasāžas veic ar tādu
ievadīšanas veidu, kas visdrīzāk var izraisīt virulences
reversiju;
30.4. vakcīnas celma bioloģiskās
īpašības. Var izrādīties nepieciešami citi testi, lai pēc
iespējas precīzāk noteiktu vakcīnas celma raksturīgās bioloģiskās
īpašības (piemēram, neirotropismu);
30.5. rekombinēšanās vai celmu
genomu pārkārtošanās. Izskata jautājumu par rekombinēšanās vai
genomu pārkārtošanās iespēju ar laukiem vai citiem celmiem.
31. Atliekvielu izpēte:
31.1. attiecībā uz imunoloģiskām
veterinārajām zālēm parasti nav nepieciešams veikt atliekvielu
izpēti. Tomēr gadījumos, ja imunoloģisku veterināro zāļu ražošanā
izmanto palīgvielas un/vai konservantus, sniedz informāciju par
iespēju, ka pārtikas produktos saglabājas atliekvielas.
Vajadzības gadījumā izpēta šādu atliekvielu iedarbību. Dzīvu
vakcīnu gadījumā pret zoonozēm papildus šī pielikuma
30.2.apakšpunktā aprakstītajiem pētījumiem var pieprasīt noteikt
atliekvielas zāļu injicēšanas vietā;
31.2. iesaka zāļu izdalīšanās
laikposmu un spriež par tā atbilstību saistībā ar atliekvielu
jebkāda veida izpēti, kas ir veikta.
32. Mijiedarbība – norāda zināmās
mijiedarbības ar citiem produktiem.
33. Lauka pētījumi– laboratorijas
pētījumu rezultātiem pievieno papildu datus no lauka pētījumiem,
ja vien nav pamatots, ka lauka pētījumi nav nepieciešami.
34. Ekotoksiskums:
34.1. imunoloģisku veterināro zāļu
ekotoksiskuma pētījuma uzdevums ir novērtēt zāļu lietošanas
iespējamo kaitīgo ietekmi uz vidi un konstatēt visus vajadzīgos
piesardzības pasākumus šā riska samazināšanai;
34.2. ekotoksiskuma novērtējums ir
obligāts jebkādam reģistrācijas iesniegumam imunoloģisku
veterināro zāļu reģistrācijai, izņemot reģistrācijas iesniegumus,
kas iesniegti atbilstoši šo noteikumu 13.9. un 13.10.apakšpunktā
un 18., 19., 20. un 21.punktā noteiktajām prasībām;
34.3. ekotoksiskuma novērtējumu
veic divos posmos atbilstoši šādiem nosacījumiem:
34.3.1. pirmajā posmā pētnieks
novērtē apjomu, kādā ir iespējama zāļu, to aktīvo vielu vai
attiecīgo metabolītu ietekme uz vidi, ņemot vērā:
34.3.1.1. mērķa sugu un piedāvāto
lietošanas modeli (piemēram, zāļu masveida lietošana vai zāļu
lietošana atsevišķam dzīvniekam),
34.3.1.2. lietošanas paņēmienu,
bet īpaši apjomu, kādā zāles tieši iekļūst vides sistēmā,
34.3.1.3. zāļu, to aktīvo vielu
vai attiecīgo metabolītu iespējamā izdalīšanās no apstrādāto
dzīvnieku organisma vidē, šādu izdalījumu saglabāšanās,
34.3.1.4. neizmantoto zāļu vai no
tām radušos atkritumu likvidēšana;
34.3.2. ja pirmā posma secinājumi
norāda uz iespējamu zāļu ietekmi uz vidi, reģistrācijas
iesnieguma iesniedzējs veic otru posmu un novērtē zāļu potenciālo
ekotoksiskumu. Šim nolūkam reģistrācijas iesnieguma iesniedzējs
nosaka apjomu un ilgumu, kādā vide ir pakļauta zāļu ietekmei, un
ziņas par fizikālajām, ķīmiskajām, farmakoloģiskajām un/vai
toksikoloģiskajām īpašībām savienojumam, kurš iegūts, veicot
citus testus un pētījumus, kas noteikti šajos noteikumos. Ja
nepieciešams, var veikt tālākus pētījumus par zāļu ietekmi
(augsne, ūdens, gaiss, ūdens organismi, sugas, kas nav mērķa
sugas);
34.4. tālākos pētījumus veic
saskaņā ar testa protokoliem, kas noteikti normatīvajos aktos par
laboratoriskajām metodēm ķīmisko vielu un ķīmisko produktu
fizikālo, ķīmisko, toksikoloģisko vai ekotoksikoloģisko īpašību
noteikšanai. Ja protokolos nav skaidri noteikts pētījumu gala
termiņš, tālākos pētījumus veic saskaņā ar citiem starptautiski
atzītiem protokoliem par imunoloģiskajām veterinārajām zālēm
un/vai aktīvajām vielām un/vai izdalītajiem metabolītiem. Testu
skaita un veida izvēle un rezultātu novērtēšanas kritēriju izvēle
ir atbilstoša zinātnes attīstības līmenim laikā, kad iesniegts
reģistrācijas iesniegums.
IV. Iedarbīguma
pētījumi
35. Šajā daļā aprakstīto pētījumu
uzdevums ir apstiprināt imunoloģisko veterināro zāļu iedarbīgumu.
Reģistrācijas iesnieguma iesniedzēja prasības par imunoloģisko
veterināro zāļu īpašībām, iedarbību un lietošanu pilnā apmērā
apstiprina speciālo pētījumu rezultāti, kuri pievienoti
reģistrācijas iesniegumam.
36. Ziņas un dokumentus, ko
pievieno reģistrācijas iesniegumam atbilstoši šo noteikumu 13.9.
un 13.10.apakšpunktā un 18., 19., 20. un 21.punktā noteiktajām
prasībām, iesniedz saskaņā ar šī pielikuma 26.punkta prasībām un
ar turpmāk minētajiem noteikumiem:
36.1. veterināros klīniskos
pētījumus veic saskaņā ar pilnībā noteiktu sīki izklāstītu
pētījumu protokolu, ko reģistrē rakstiski pirms pētījuma
sākšanas. Lai nodrošinātu dzīvnieku labturības prasību
ievērošanu, pētījumos iesaistītie dzīvnieki ir pakļauti
veterinārajai uzraudzībai. Dzīvnieku labturības prasības pilnībā
ievēro, sagatavojot pētījumu protokolu un veicot pētījumu;
36.2. obligāti nepieciešamas
iepriekš izveidotas sistēmiskas rakstiskas procedūras klīnisko
pētījumu organizēšanai, veikšanai, datu vākšanai, dokumentēšanai
un apstiprināšanai;
36.3. pirms pētījuma uzsākšanas
saņem pētījumā izmantojamo dzīvnieku īpašnieka piekrišanu un to
dokumentē. Dzīvnieka īpašnieku rakstveidā informē par sekām, kas
pēc piedalīšanās pētījumā var rasties, apstrādāto dzīvnieku
likvidējot vai iegūstot pārtikas produktus no apstrādātā
dzīvnieka. Šī paziņojuma kopiju, ko parakstījis un datējis
dzīvnieka īpašnieks, iekļauj pētījuma dokumentācijā;
36.4. ja vien veterināros
klīniskos pētījumus neveic pēc aklās metodes, marķēšanas
nosacījumus, kas noteikti normatīvajos aktos par veterināro zāļu
marķēšanas, izplatīšanas un kontroles nosacījumiem, piemēro
veterinārajos klīniskajos pētījumos izmantojamo zāļu marķējumam.
Marķējumā skaidri un nenodzēšami norāda frāzi “Lietošanai vienīgi
klīniskos veterinārajos pētījumos”.
37. Vakcīnas celmu izvēli pamato,
balstoties uz epizooloģiskajiem datiem.
38. Laboratorijā veiktie
iedarbības pētījumi ir kontrolētie pētījumi, ietverot
neapstrādātus kontroles dzīvniekus, atbilstoši šādiem
nosacījumiem:
38.1. šos pētījumus papildina
lauka pētījumi, ietverot neapstrādātus kontroles dzīvniekus;
38.2. visus pētījumus apraksta
pietiekami sīki, lai tos var atkārtot kontroles pētījumos, ko
veic pēc Zāļu valsts aģentūras pieprasījuma. Pētnieks uzrāda
izmantoto metodiku apstiprinājumu. Rezultātus atspoguļo
precīzi;
38.3. ziņo par iegūtajiem
rezultātiem neatkarīgi no to iznākuma (vai tie ir labvēlīgi vai
nelabvēlīgi).
39. Imunoloģisko veterināro zāļu
iedarbīgumu uzrāda katrai dzīvnieku sugai un katrai dzīvnieku
kategorijai, kura ieteikta vakcinēšanai, ar katru ieteikto
ievadīšanas veidu un izmantojot piedāvāto lietošanas grafiku.
Atbilstīgi novērtē vakcīnas ietekmi kā pasīvi iegūto vai no mātes
iedzimto antivielu ietekmi. Informāciju par pazīmju sākumu un
aizsardzības ilgumu apstiprina ar pētījumu datiem.
40. Multivalentām un kombinētām
imunoloģiskām veterinārajām zālēm uzrāda katras sastāvdaļas
iedarbību. Ja šīs zāles iesaka lietošanai savienojumā ar vai
vienlaikus ar citām veterinārām zālēm, uzrāda zāļu
saderīgumu.
41. Ja zāles veido daļu no
vakcinācijas sistēmas, ko iesaka reģistrācijas iesnieguma
iesniedzējs, jāatspoguļo zāļu pirmatnējā jeb aktivizējošā
iedarbība vai zāļu ieguldījums vakcinācijas sistēmas kopējā
iedarbībā.
42. Izmantojamā zāļu deva ir
daudzums, ko ieteic lietošanai un kas satur maksimālo titru vai
potenci, par ko ir iesniegts reģistrācijas iesniegums.
43. Paraugus, ko izmanto
iedarbīguma pētījumiem, ņem no sērijas vai sērijām, kas saražotas
ražošanas procesā, kas atbilst reģistrācijas iesniegumā
noteiktajam.
44. Diagnosticēšanai paredzētajām
imunoloģiskām veterinārajām zālēm reģistrācijas iesnieguma
iesniedzējs norāda, kā interpretējama apstrādāto dzīvnieku
reakcija uz šīm zālēm.
45. Laboratoriskie pētījumi:
45.1. imunoloģisko veterināro zāļu
iedarbīgumu demonstrē labi kontrolētos laboratorijas apstākļos ar
reakciju, ko mērķa dzīvniekā provocē imunoloģisku veterināro zāļu
ievadīšana ieteiktajos lietošanas apstākļos. Ciktāl iespējams,
apstākļi, kuros veic provokāciju, atveido dabīgos inficēšanās
apstākļus, piemēram, ņemot vērā provokācijas organisma apjomu un
provokācijas ievadīšanas veidu;
45.2. ja iespējams, uzskaita un
dokumentāli apraksta imunitātes mehānismu (šūnu– vides/humorālo,
lokālās/vispārējās imunglobulīnu klases), kas tiek ierosināts pēc
imunoloģisko veterināro zāļu ievadīšanas mērķa dzīvniekiem ar
ieteikto ievadīšanas veidu.
46. Lauka pētījumi:
46.1. laboratorijas pētījumu
rezultātiem pievieno papildu datus no lauka pētījumiem, ja vien
nav pamatots, ka lauka pētījumi nav nepieciešami;
46.2. ja laboratorijas pētījumi
nevar pamatot iedarbīguma aspektu, atļauts izmantot tikai lauka
pētījumus.
V. Ziņas un
dokumenti par drošuma testēšanu un iedarbīguma izmēģinājumiem
imunoloģiskām veterinārajām zālēm
47. Dokumentācijā par drošuma un
iedarbīguma pētījumiem, kā jebkurā zinātniskā darbā, iekļauj
ievadu, kur definē aplūkojamo objektu, un norāda testus, kas
veikti saskaņā ar šī pielikuma III un IV daļas noteikumiem, ar
kopsavilkumu, ar atsaucēm uz izmantoto literatūru. Norāda un
argumentē, ja ir izlaists kāds šī pielikuma III un IV daļā
noteiktais tests vai pētījumi.
48. Par laboratorijas pētījumiem
sniedz šādas ziņas:
48.1. kopsavilkumu;
48.2. tās iestādes nosaukumu, kas
veic pētījumus;
48.3. sīki izklāstītu eksperimenta
protokolu, kurā norāda:
48.3.1. izmantoto metožu,
aparatūras un materiālu aprakstu,
48.3.2. sīki izklāstītas ziņas par
dzīvnieku sugu, šķirni vai līniju, dzīvnieku kategorijām, ziņām
par dzīvnieku iegūšanu, to identifikāciju un skaitu,
48.3.3. dzīvnieku izmitināšanas
apstākļus un barošanas nosacījumus (inter alia nosakot,
vai tie ir brīvi no jebkādiem specifiskiem patogēniem un/vai
specifiskām antivielām, dzīvnieku barībā esošu piedevu īpašībām
un kvantitāti),
48.3.4. lietoto zāļu devu,
ievadīšanas veidu, grafiku un datumus,
48.3.5. izmantoto statistikas
metožu aprakstu;
48.4. kontroles dzīvniekiem– vai
tie ir saņēmuši placebo vai nav saņēmuši nekādu ārstēšanu;
48.5. vispārējie un individuālie
novērojumi un iegūtie rezultāti (ar vidējām un standarta
novirzēm), gan labvēlīgi, gan nelabvēlīgi. Datus apraksta
pietiekami sīki, lai rezultātus var izvērtēt kritiski,
neizmantojot autora interpretāciju. Neapstrādātus datus atspoguļo
tabulas veidā. Rezultātus skaidrojot un ilustrējot, tos var
papildināt ar ierakstu atskaņošanu, mikrofilmu fotogrāfijām
u.tml.;
48.6. novēroto blakusparādību
īpašības, biežums un ilgums;
48.7. to dzīvnieku skaits, kas
pirms laika izņemti no pētījumiem, un šādas izņemšanas
iemeslus;
48.8. rezultātu statistiskā
analīze, ja tādu pieprasa testa programma, un datu
variablums;
48.9. jebkādas papildu slimības
sastopamības un norises gaita;
48.10. ziņas par zālēm (kas nav
pētāmās zāles), kuru lietošana bija nepieciešama pētījuma
gaitā;
48.11. objektīvs iztirzājums par
iegūtajiem rezultātiem, kas ļauj izdarīt secinājumus par produkta
drošumu un iedarbīgumu.
49. Lauka pētījumi. Ziņas par
lauka studijām pietiekami sīki izklāstītas, kas ļauj veidot
objektīvu spriedumu. Ziņās par lauka studijām iekļauj šādu
informāciju:
49.1. kopsavilkumu;
49.2. atbildīgā pētnieka uzvārdu,
adresi, amatu un kvalifikāciju;
49.3. lietošanas vietu un laiku,
dzīvnieka īpašnieka uzvārdu un adresi;
49.4. sīkas ziņas par pētījuma
protokolu, kas apraksta izmantotās metodes, aparatūru un
materiālus, ziņas par ievadīšanas veidu, lietošanas grafiku, devu
un dzīvnieku kategorijām, novērojumu ilgumu, seroloģisko reakciju
un citiem izmeklējumiem, kas veikti dzīvniekiem pēc zāļu
ievadīšanas;
49.5. kontroles dzīvniekiem– vai
tie ir saņēmuši placebo vai nav saņēmuši nekādu
ārstēšanu;
49.6. apstrādāto un kontroles
dzīvnieku identifikācija (kolektīvi vai individuāli): suga,
šķirne vai līnijas, vecums, svars, dzimums, fizioloģiskais
stāvoklis;
49.7. audzēšanas un barošanas
metodes īss aprakstu, kurā nosaka barības piedevu veidu un
daudzumu;
49.8. visas ziņas par novērotajiem
faktiem, novērtējumu un rezultātu (ar vidējo un standarta
novirzi). Individuālos datus norāda, ja indivīdiem veic testus un
mērījumus;
49.9. klīnisko pētījumu rezultātus
neatkarīgi no tā, vai tie ir labvēlīgi vai nelabvēlīgi, pilnā
apmērā nosakot novērojumu un aktivitātes objektīvo testu
rezultātus, kas nepieciešami izstrādājuma novērtēšanai. Uzskaita
izmantotās metodes un izskaidro nozīmīgumu, kāds piemīt jebkādiem
variantiem rezultātos;
49.10. ietekme uz dzīvnieku kopējo
vērtību (piemēram, dējību, pienīgumu un reproduktīvajām
funkcijām);
49.11. to dzīvnieku skaitu, kas
pirms laika izņemti no pētījumiem, un šādas izņemšanas
iemeslus;
49.12. novēroto blakusparādību
īpašības, biežumu un ilgumu;
49.13. papildu slimības
sastopamību un norises gaitu;
49.14. visas ziņas par
veterinārajām zālēm (izņemot tās, kam tiek veikts pētījums), kas
var būt lietotas pirms testa zālēm vai vienlaikus ar tām, vai
novērošanas laikposmā. Ziņas par novērotu mijiedarbību;
49.15. objektīvu iztirzājumu par
iegūtajiem rezultātiem, kas ļauj izdarīt secinājumus par produkta
drošumu un iedarbīgumu.
50. Vispārīgs atzinums. Sniedz
vispārīgu atzinumu par pētījumu un testu rezultātiem, kas veikti
atbilstoši šī pielikuma III un IV daļas prasībām. Tajos iekļauj
objektīvu iztirzājumu par iegūtajiem rezultātiem, kas ļauj
izdarīt secinājumus par imunoloģisku veterināro zāļu drošumu un
iedarbīgumu.
51. Bibliogrāfiskās atsauces. Sīki
uzskaita bibliogrāfiskās atsauces, kas citētas šī pielikuma
47.punktā minētājā kopsavilkumā.
Zemkopības ministra vietā –
iekšlietu ministrs Dz.Jaundžeikars
Zemkopības
ministrijas iesniegtajā redakcijā
4.pielikums
Ministru kabineta
2006.gada 18.jūlija noteikumiem
Nr.600
Krāsvielas, ko
atļauts pievienot veterinārajām zālēm
|
1
|
2
|
3
|
4
|
|
Nr. p.k.
|
Starptautiskais (E) Nr.
|
Nosaukums angļu valodā
|
Nosaukums latviešu valodā
|
|
1
|
2
|
3
|
4
|
|
1.
|
E 100
|
Curcumin
|
Kurkumīns
|
|
2.
|
E 101
|
Lactoflavin (Riboflavin)
|
Riboflavīns
|
|
3.
|
E 102
|
Tartrazine
|
Tartrazīns
|
|
4.
|
E 104
|
Quinoline yellow
|
Hinolīna dzeltenais
|
|
5.
|
E 110
|
Orange yellow S
Sunset yellow FCF
|
Oranždzeltenais S
Saulrieta dzeltenais FCF
|
|
6.
|
E 120
|
Cochineal
Carminic acid
|
Košenile
Karmīnskābe
|
|
7.
|
E 122
|
Azorubine
Carmoisine
|
Azorubīns
Karmoizīns
|
|
8.
|
E 123
|
Amaranth
|
Amarants
|
|
9.
|
E 124
|
Cochineal Red A
Ponceau 4R
|
Košenilsarkanais A
Kumačs 4R
|
|
10.
|
E 127
|
Erythrosine
|
Eritrozīns
|
|
11.
|
E 131
|
Patent Blue V
|
Patentzilais V
|
|
12.
|
E 132
|
Indigotin (indigo carmine)
|
Indigotīns (indigokarmīns)
|
|
13.
|
E 140
|
Chlorophylls
|
Hlorofili
|
|
14.
|
E 141
|
Copper complexes of
chlorophylls and chlorophyllins
|
Hlorofilu un hlorofilīnu vara
kompleksi
|
|
15.
|
E 142
|
Acid brilliant green BS
(lissamine green)
|
Skābais briljantzaļais BS
(lizamīnzaļais)
|
|
16.
|
E 150
|
Caramel
|
Karamele
|
|
17.
|
E 151
|
Brilliant Black BN
Black PN
|
Briljantmelnais BN
Melnais PN
|
|
18.
|
E 153
|
Carbo medicinalis vegetalis
(charcoal)
|
Medicīniskā ogle (kokogle)
|
|
19.
|
E 160
|
Carotenoids:
|
Karotinoīdi:
|
|
19.1.
|
E 160a
|
Alpha-, beta-,
gamma-carotene
|
Alfa-, beta-, gamma-
karotīns
|
|
19.2.
|
E 160b
|
Bixin
Norbixin (Roucou Annatto)
|
Biksīns
Norbiksīns
|
|
19.3.
|
E 160c
|
Capsanthin
Capsorubin
|
Kapsantīns
Kapsorubīns
|
|
19.4.
|
E 160d
|
Lycopene
|
Likopēns
|
|
19.5.
|
E 160e
|
Beta-apo-8’carotenal (C30)
|
Beta-apo-8-karotināls (C30)
|
|
19.6.
|
E 160f
|
Ethyl ester of beta-apo-8’
carotenoic acid (C30)
|
Beta-apo-8-karotīnskābes
etilesteris (C30)
|
|
20.
|
E 161
|
Xanthophylls:
|
Ksantofili:
|
|
20.1.
|
E 161a
|
Flavoxanthin
|
Flavoksantīns
|
|
20.2.
|
E 161b
|
Lutein
|
Luteīns
|
|
20.3.
|
E 161c
|
Kryptoxanthin
|
Kriptoksantīns
|
|
20.4.
|
E 161d
|
Rubixanthin
|
Rubiksantīns
|
|
20.5.
|
E 161e
|
Violoxanthin
|
Violoksantīns
|
|
20.6.
|
E 161f
|
Rhodoxant
|
Rodoksants
|
|
20.7.
|
E 161g
|
Canthaxanthin
|
Kantaksantīns
|
|
21.
|
E 162
|
Beetroot red
Betanin
|
Biešu sarkanais
Betanīns
|
|
22.
|
E 163
|
Anthocianins
|
Antocianīni
|
|
23.
|
E 170
|
Calcium carbonate
|
Kalcija karbonāts
|
|
24.
|
E 171
|
Titanium dioxide
|
Titāna dioksīds
|
|
25.
|
E 172
|
Iron oxides and hydroxides
|
Dzelzs oksīdi un hidroksīdi
|
|
26.
|
E 173
|
Aluminium
|
Alumīnijs
|
|
27.
|
E 174
|
Silver
|
Sudrabs
|
|
28.
|
E 175
|
Gold
|
Zelts
|
Piezīmes.
1)Vispārīgie un specifiskie
tīrības kritēriji krāsvielām, kuras atļauts pievienot zālēm, kā
arī analīzes metodes, ko lieto, lai noteiktu krāsvielu atbilstību
minētajiem kritērijiem, ir identiskas tām, kādas piemēro pārtikas
krāsvielām.
2)Eksportam paredzētām zālēm var
būt pievienotas citas krāsvielas, ja šo krāsvielu izmantošana ir
atļauta importētājvalstī.
Zemkopības ministra vietā –
iekšlietu ministrs Dz.Jaundžeikars
Zemkopības
ministrijas iesniegtajā redakcijā
5.pielikums
Ministru kabineta
2006.gada 18.jūlija noteikumiem
Nr.600
Veterināro zāļu
reģistrācijas paplašināšana
Izmaiņas reģistrācijas
dokumentācijā, par ko zāļu reģistrācijas īpašnieks iesniedz
iesniegumu reģistrācijas paplašināšanai, ir šādas:
1. Izmaiņas, kas saistītas ar
aktīvo(–ajām) vielu(–ām):
1.1. aktīvās(–o) vielas(–u)
aizstāšana ar citu sāls vai estera kompleksu vai to atvasinājumu
(ar to pašu ārstniecisko sastāvdaļu), ja iedarbības un drošības
raksturlielumi būtiski neatšķiras;
1.2. aktīvās vielas aizstāšana ar
citu izomēru vai citu izomēru maisījumu, maisījuma aizstāšana ar
izolētu izomēru (piemēram, racemātu aizstāj ar vienu
enantiomēru), ja iedarbības un drošības raksturlielumi būtiski
neatšķiras;
1.3. bioloģiska vielas vai
biotehnoloģiska produkta aizstāšana ar vielu, kuras molekulai ir
nedaudz atšķirīga struktūra. Vektora pārveidošana, kuru izmanto
antigēna, kā arī izejmateriāla izgatavošanā (arī jauna izejas
(galveno) šūnu banka no cita avota), ja iedarbības un drošuma
raksturlielumi būtiski neatšķiras;
1.4. jauns ligands vai savienojuma
mehānisms radiofarmaceitiskam preparātam;
1.5. izmaiņas ekstraģenta
šķīdinātāja vai augu izcelsmes zāļu augu attiecībā, ja iedarbības
un drošuma raksturlielumi būtiski neatšķiras.
2. Izmaiņas veterināro zāļu
stiprumā, formā vai lietošanas veidā:
2.1. izmaiņas biopieejamībā;
2.2. izmaiņas farmakokinētikā
(piemēram, atbrīvošanās ātruma pakāpē);
2.3. izmaiņas vai jauna stipruma,
kā arī iedarbības pievienošana;
2.4. izmaiņas vai jaunas zāļu
formas pievienošana;
2.5. lietošanas veida izmaiņas vai
jauna veida pievienošana. Ja zāles ievada parenterāli, norāda
ievadīšanas veidu (piemēram, intraarteriāli, intravenozi,
intramuskulāri, zemādas. Mājputniem atbilstošs ievadīšanas veids
ir vakcinēšanai izmantota respiratora, orāla un okulāra
ievadīšana).
3.Citas veterinārajām zālēm, kas
paredzētas produktīvajiem dzīvniekiem, raksturīgas izmaiņas:
mērķa sugas maiņa vai mērķa sugas pievienošana.
Zemkopības ministra vietā –
iekšlietu ministrs Dz.Jaundžeikars
Zemkopības
ministrijas iesniegtajā redakcijā
6.pielikums
Ministru kabineta
2006.gada 18.jūlija noteikumiem
Nr.600
Nelielas IA un
IB tipa izmaiņas
Nacionālā reģistrācijas procedūrā
reģistrētām veterinārām zālēm (neattiecas uz veterinārām zālēm,
ko reģistrē savstarpējā atzīšanas procedūrā un decentralizētā
procedūrā) nelielas IA un IB tipa izmaiņas ir šādas:
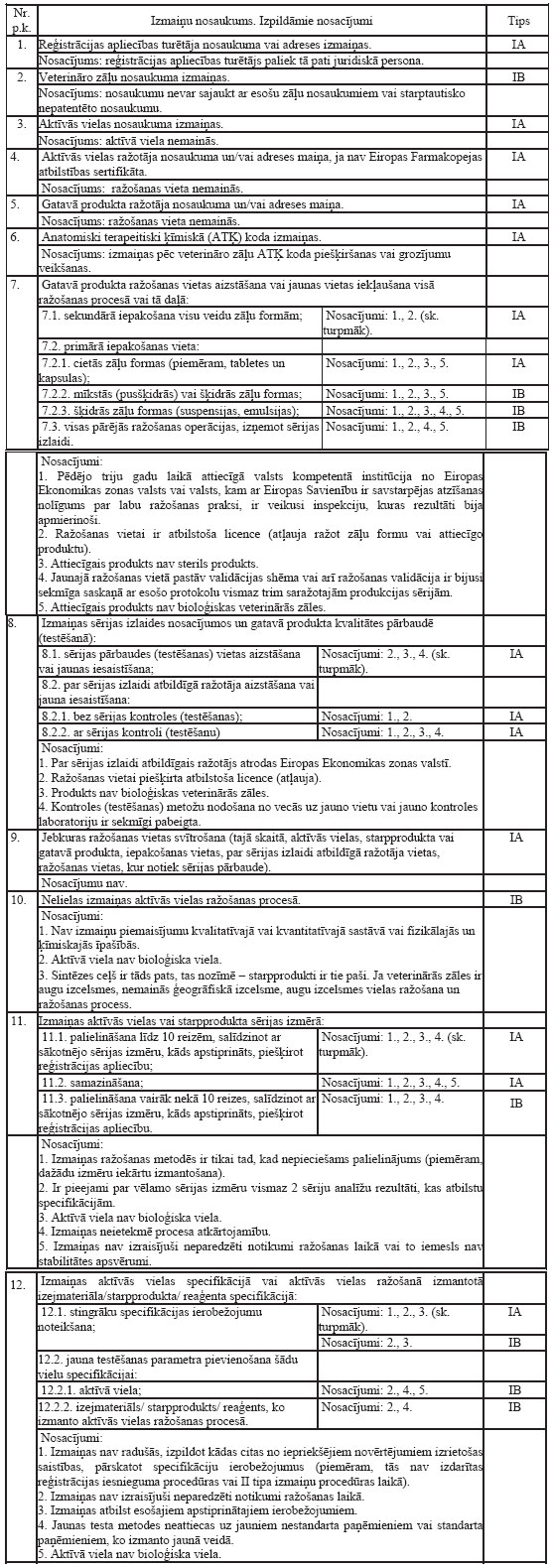
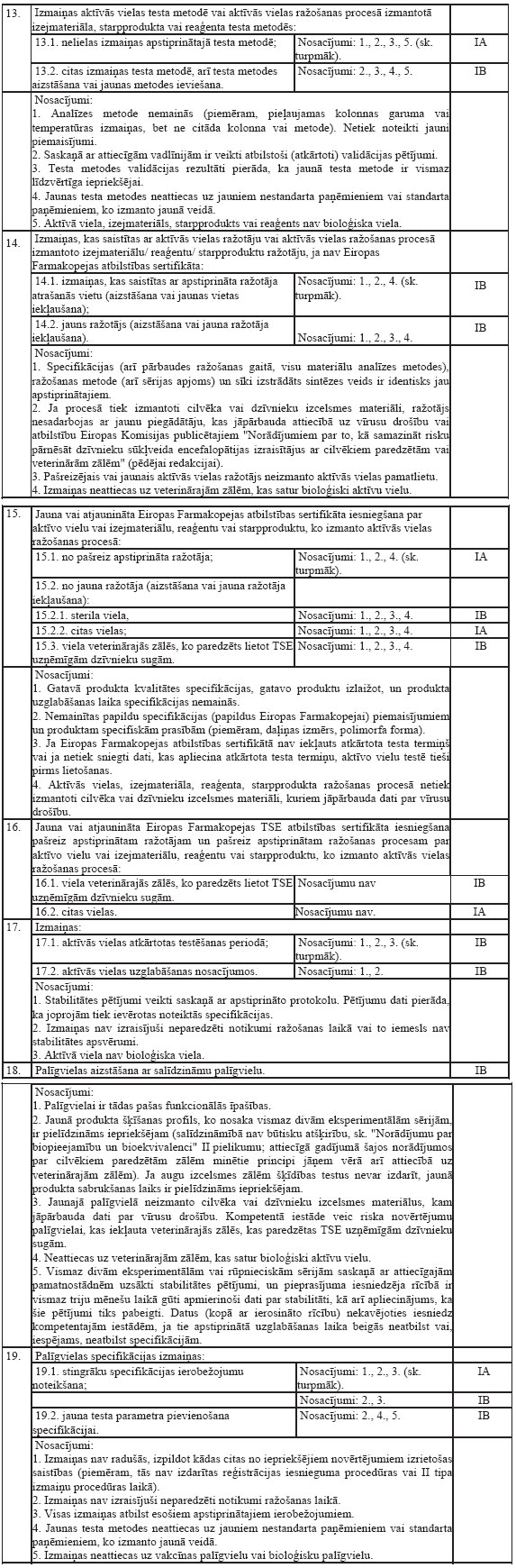
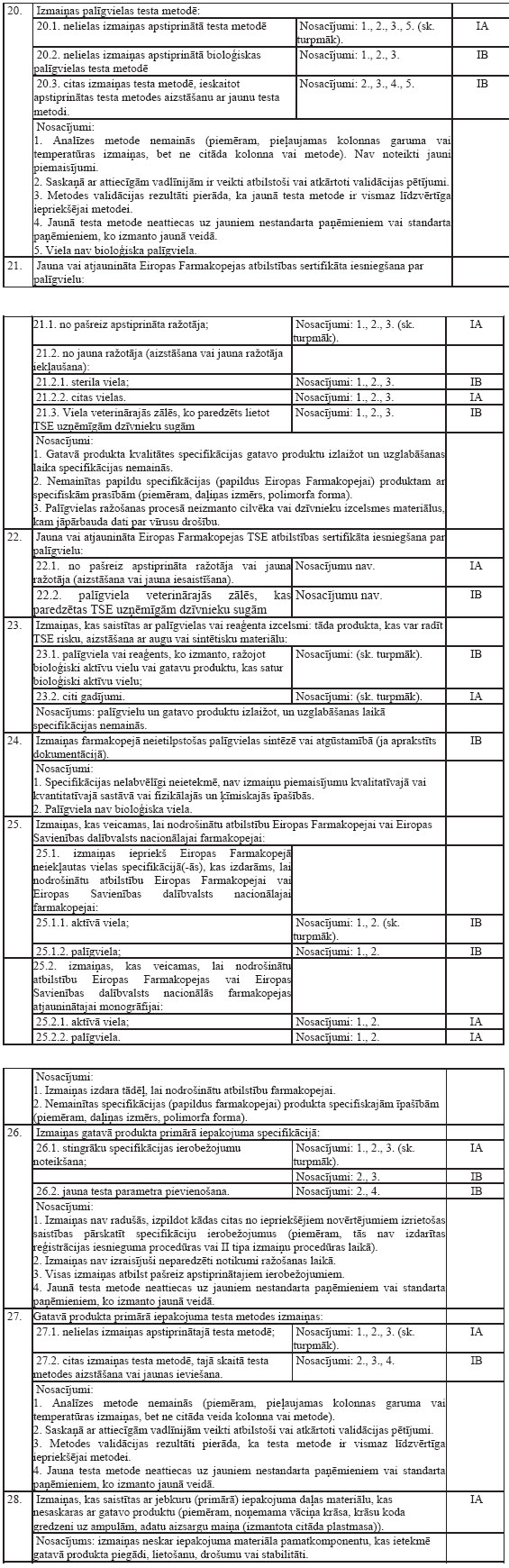
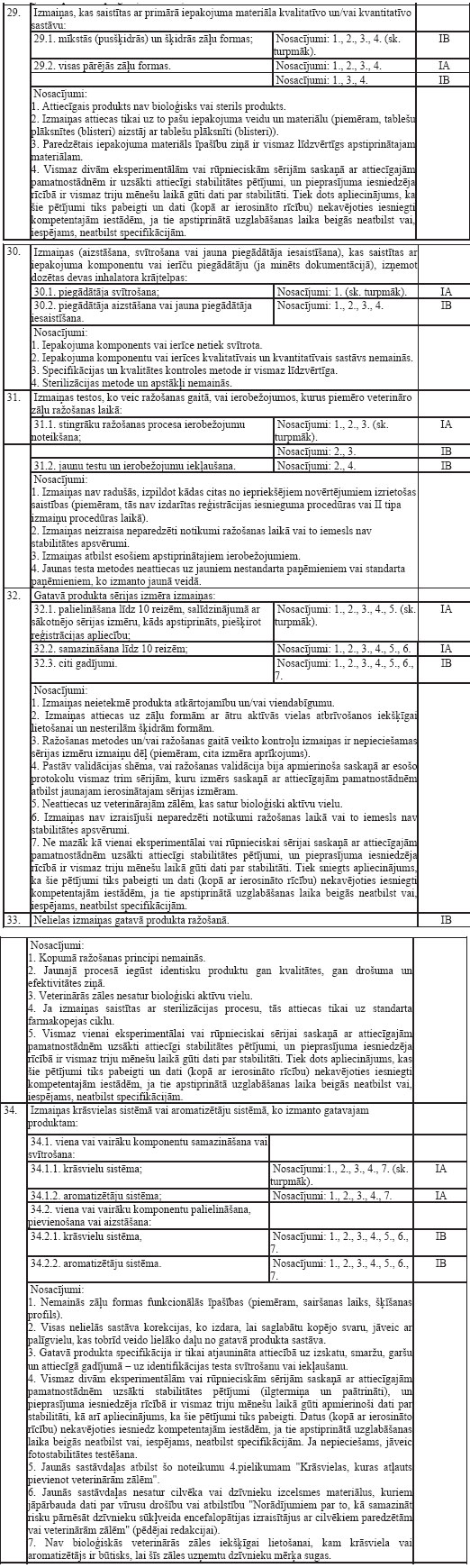
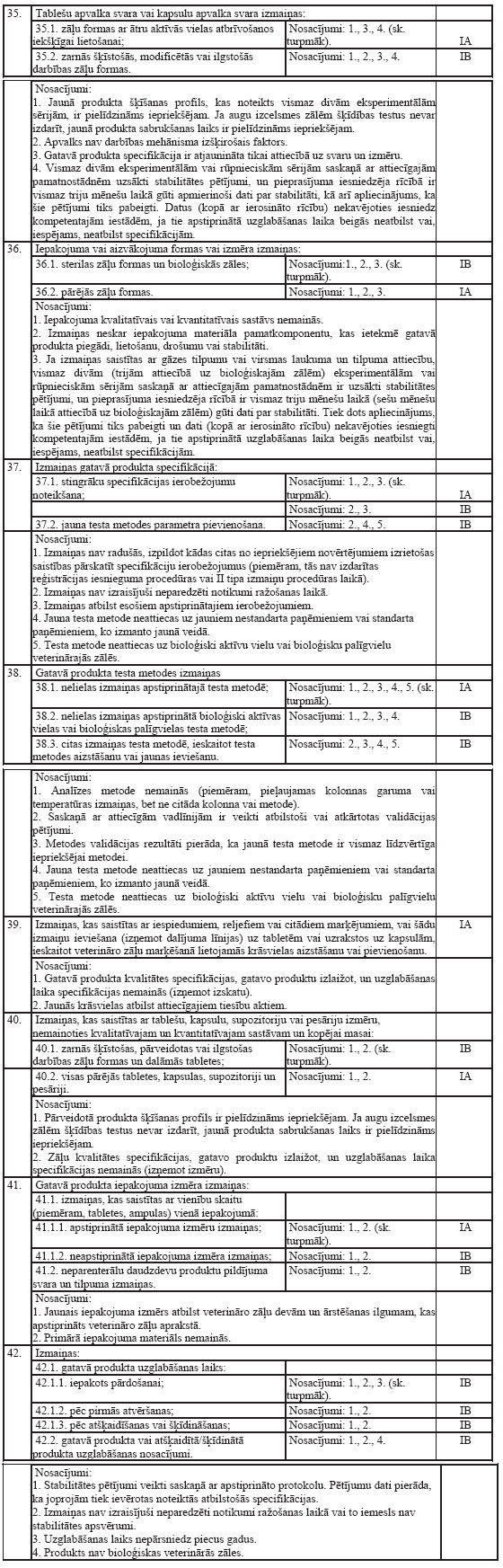
Piezīmes.
1) Nosacījumi, kas jāizpilda par
konkrētajām izmaiņām, lai tiktu ievērotas IA tipa vai IB tipa
procedūras, norādīti katrai apakškategorijai un uzskaitīti aiz
katrām izmaiņām.
2) Vienlaicīgi jāiesniedz
pieprasījums izmaiņu apstiprināšanai par visām izrietošajām vai
paralēlajām izmaiņām, kas var būt saistītas ar izmaiņām, par
kurām iesniegts pieprasījums izmaiņu apstiprināšanai, skaidri
norādot saistību starp šīm izmaiņām.
3) Paziņojumam, kas ietver Eiropas
Farmakopejas atbilstības sertifikātu, un gadījumos, kad izmaiņas
attiecas uz dokumentāciju, kas iesniegta šī sertifikāta
iegūšanai, izmaiņām nepieciešamie dokumenti jāiesniedz Eiropas
Zāļu kvalitātes direktorātā (EDQM). Ja pēc šo izmaiņu
novērtēšanas sertifikātu pārstrādā, atjaunina visas attiecīgās
reģistrācijas apliecības (reģistrācijas dokumentācija). Daudzos
gadījumos to var izdarīt, paziņojot par IA tipa izmaiņām.
4) Bioloģiskās veterinārās zāles
ir preparāts, kurā aktīvā viela ir bioloģiska viela. Bioloģisku
vielu ražo vai ekstrahē no bioloģiska avota, un tās apraksta un
kvalitātes noteikšanai veic fizikālus, ķīmiskus un bioloģiskus
testus, kā arī kontrolē ražošanas procesu. Par bioloģiskajām
veterinārajām zālēm uzskata imunoloģiskās veterinārās zāles.
5) Izmaiņas olbaltumvielas
nesaturoša komponenta ražošanas procesā biotehnoloģijas posma
ieviešanas dēļ var izdarīt saskaņā ar I tipa izmaiņu 15. vai
20.noteikumu. Šīs konkrētās izmaiņas neskar citas šajā pielikumā
uzskaitītās izmaiņas, ko var piemērot konkrētajā gadījumā.
Minētais neattiecas uz biotehnoloģiskā procesā iegūtu
olbaltumvielas saturošu komponentu ieviešanu zāļu sastāvā
centralizētai reģistrācijai pakļautajām zālēm, uz ko attiecas
Eiropas Komisijas 2003.gada 3.jūnija Regula (EK) Nr.1085/2003 par
izmaiņu izskatīšanu cilvēkiem paredzētu zāļu un veterināro zāļu
tirdzniecības atļaujas nosacījumos, uz ko attiecas Regula, ar ko
nosaka Kopienas procedūru tam, kā apstiprināt un pārraudzīt
zāles, kuras paredzētas izmantošanai cilvēkiem un veterinārijā,
un ar ko nodibina Eiropas Zāļu aģentūru.
6) Zāļu valsts aģentūrai nav
jāpaziņo par atjauninātu Eiropas Farmakopejas vai Eiropas
Savienības dalībvalsts farmakopejas monogrāfiju, ja atbilstība
atjauninātajai monogrāfijai tiek sagatavota sešu mēnešu laikā no
tās publicēšanas un reģistrētu veterināro zāļu dokumentācijā ir
atsauce uz esošo redakciju.
7) Šajā pielikumā minētā testa
metode nozīmē to pašu, ko analītiskā metode, un ierobežojumi
nozīmē to pašu, ko pieņemšanas kritēriji.
Zemkopības ministra vietā –
iekšlietu ministrs Dz.Jaundžeikars
