ATSAUCĒ IETVERT:
2013/129.6
RĪKI
Ministru kabineta noteikumi: Šajā laidienā 8 Pēdējās nedēļas laikā 20 Visi
Ministru kabineta noteikumi Nr.354
Rīgā 2013.gada 2.jūlijā (prot. Nr.37 16.§)
Kosmētikas līdzekļu būtisko prasību nodrošināšanas kārtība
Izdoti saskaņā ar likuma "Par atbilstības novērtēšanu" 7.panta
pirmo daļu
un Patērētāju tiesību aizsardzības likuma 21.panta pirmo daļu
1. Noteikumi nosaka kosmētikas līdzekļu būtiskās prasības un to ievērošanas uzraudzības mehānismu, kā arī to kosmētikas līdzekļu marķēšanas kārtību, kuriem nepieciešams sevišķs marķējums.
2. Juridiskā vai fiziskā persona, kas laiž tirgū vai dara pieejamus tirgū kosmētikas līdzekļus, ievēro Eiropas Parlamenta un Padomes 2009.gada 30.novembra Regulā (EK) Nr.1223/2009 par kosmētikas līdzekļiem (turpmāk – regula Nr.1223/2009) noteiktās prasības.
3. Veselības inspekcija ir kompetentā iestāde saskaņā ar regulas Nr.1223/2009 34.panta pirmo punktu (turpmāk – kompetentā iestāde), un tā nodrošina tirgus iekšējo kontroli.
4. Sabiedrības ar ierobežotu atbildību "Rīgas Austrumu klīniskā universitātes slimnīca" Toksikoloģijas un sepses klīnikas Saindēšanās informācijas centrs elektroniskā veidā saņem no Eiropas Komisijas regulas Nr.1223/2009 13.panta 6.punktā minēto informāciju par kosmētikas līdzekli.
5. Regulā Nr.1223/2009 noteiktās atbildīgās personas un izplatītāji izmanto šo noteikumu 1.pielikumā norādīto veidlapu, lai atbilstoši regulas Nr.1223/2009 23.pantam paziņotu kompetentajai iestādei par būtisku nevēlamu kosmētikas līdzekļa ietekmi.
6. Ja ārstniecības persona vai gala patērētājs ziņo kompetentajai iestādei par būtisku nevēlamu kosmētikas līdzekļa ietekmi, ziņojumā sniedz šo noteikumu 1.pielikumā minēto informāciju.
7. Lai nodrošinātu kosmētikas līdzekļa būtisko prasību ievērošanu un noteiktu tā sastāvā šo noteikumu 2.pielikumā minētās vielas, ņemot un sagatavojot paraugus, izmanto šo noteikumu 2.pielikumā minētās metodes.
8. Šo noteikumu 7.punktā un regulas Nr.1223/2009 1.pielikumā minētos testus veic laboratorijas, kas atbilst normatīvo aktu prasībām par laboratoriju darba kvalitāti un laboratoriju inspicēšanu, vai laboratorijas, kas akreditētas sabiedrības ar ierobežotu atbildību "Standartizācijas, akreditācijas un metroloģijas centrs" Latvijas Nacionālajā akreditācijas birojā atbilstoši standartam LVS EN ISO/IEC 17025:2005 "Testēšanas un kalibrēšanas laboratoriju kompetences vispārīgās prasības" un par kurām publicēta informācija akreditācijas biroja tīmekļa vietnē.
9. Kosmētikas līdzekļa atbilstību mikrobioloģiskās tīrības kritērijiem, kas minēti šo noteikumu 3.pielikumā, nodrošina atbildīgā persona. Kosmētikas līdzekļa mikrobioloģisko testēšanu veic saskaņā ar regulas Nr.1223/2009 12.pantā noteiktajām prasībām.
10. Latvijā ražotiem, importētiem vai izplatītiem kosmētikas līdzekļiem valsts valodā norāda regulas Nr.1223/2009 19.panta 1.punkta "b", "c", "d" un "f" apakšpunktā un 2., 3. un 4.punktā minēto informāciju.
11. Kosmētikas līdzekļiem, kas nav fasēti vai tiek fasēti pārdošanas vietā pēc pircēja pieprasījuma, vai tiek fasēti tūlītējai pārdošanai, regulas Nr.1223/2009 19.panta 1.punktā minēto informāciju norāda kosmētikas līdzeklim pievienotajā anotācijā vai etiķetē.
12. Atbilstoši regulas Nr.1223/2009 19.panta 4.punktā minētajiem nosacījumiem ražotāji, importētāji un izplatītāji nodrošina un pēc pieprasījuma spēj sniegt visu ar attiecīgo kosmētikas līdzekli saistīto informāciju (ieskaitot partijas numuru) par tiem kosmētikas līdzekļiem, kas nav fasēti vai tiek fasēti pārdošanas vietā pēc pircēja pieprasījuma, vai tiek fasēti tūlītējai pārdošanai.
13. Veselības inspekcija izsniedz kosmētikas līdzekļu ražotājiem Latvijā brīvās tirdzniecības sertifikātu, kas apliecina, ka komersants ir paziņojis par Latvijā ražotu kosmētikas līdzekļu laišanu Latvijas tirgū. Brīvās tirdzniecības sertifikāta saņemšanai kosmētikas līdzekļu ražotājs iesniedz Veselības inspekcijā iesniegumu (4.pielikums) papīra vai elektroniska dokumenta formā. Ja iesniegumu iesniedz papīra dokumenta formā, iesniegumam pievieno arī tā elektronisko formu.
14. Kompetentā iestāde uztur kosmētikas līdzekļu sastāvdaļu datubāzi, kas izveidota līdz šo noteikumu spēkā stāšanās dienai, un, ja nepieciešams, ārstniecības personām nodrošina pieeju informācijai saskaņā ar regulas Nr.1223/2009 38.panta ceturto daļu.
15. Atzīt par spēku zaudējušiem Ministru kabineta 2004.gada 20.aprīļa noteikumus Nr.354 "Noteikumi par būtiskajām prasībām kosmētikas līdzekļiem un to uzraudzības kārtību" (Latvijas Vēstnesis, 2004, 68., 185.nr.; 2005, 8., 63., 157., 201.nr.; 2006, 90., 149.nr.; 2007, 33., 108., 160.nr.; 2008, 29., 122.nr.; 2009, 29., 85., 154.nr.; 2010, 16., 68.nr.; 2011, 78., 203.nr.; 2012, 169.nr.; 2013, 44.nr.).
16. Noteikumi stājas spēkā 2013.gada 11.jūlijā.
17. Šo noteikumu 14.punkts ir spēkā līdz 2020.gada 11.jūlijam.
Informatīva atsauce uz Eiropas Savienības direktīvām
Noteikumos iekļautas tiesību normas, kas izriet no:
1) Komisijas 1980.gada 22.decembra Pirmās direktīvas 80/1335/EEK par dalībvalstu tiesību aktu tuvināšanu attiecībā uz analīzes metodēm, kas vajadzīgas, lai pārbaudītu kosmētikas līdzekļu sastāvu;
2) Komisijas 1982.gada 14.maija Otrās direktīvas 82/434/EEK par dalībvalstu tiesību aktu tuvināšanu attiecībā uz analīzes metodēm, kas vajadzīgas, lai pārbaudītu kosmētikas līdzekļu sastāvu;
3) Komisijas 1983.gada 27.septembra Trešās direktīvas 83/514/EEK par dalībvalstu tiesību aktu tuvināšanu attiecībā uz analīzes metodēm, kuras vajadzīgas, lai pārbaudītu kosmētikas līdzekļu sastāvu;
4) Komisijas 1985.gada 11.oktobra Ceturtās direktīvas 85/490/EEK par dalībvalstu tiesību aktu tuvināšanu attiecībā uz analīzes metodēm, kuras vajadzīgas, lai pārbaudītu kosmētikas līdzekļu sastāvu;
5) Komisijas 1993.gada 9.septembra Piektās direktīvas 93/73/EEK par analīzes metodēm, kas vajadzīgas, lai pārbaudītu kosmētikas līdzekļu sastāvu;
6) Komisijas 1995.gada 7.jūlija Sestās direktīvas 95/32/EK attiecībā uz analīzes metodēm, kas vajadzīgas, lai pārbaudītu kosmētikas līdzekļu sastāvu;
7) Komisijas 1996.gada 2.jūlija Septītās direktīvas 96/45/EK attiecībā uz analīzes metodēm, kas vajadzīgas, lai pārbaudītu kosmētikas līdzekļu sastāvu;
8) Komisijas 1990.gada 4.aprīļa Direktīvas 90/207/EEK, ar kuru groza Otro direktīvu 82/434/EEK par dalībvalstu tiesību aktu tuvināšanu attiecībā uz analīzes metodēm, kas vajadzīgas, lai pārbaudītu kosmētikas līdzekļu sastāvu.
Ministru prezidents Valdis Dombrovskis
Veselības ministre Ingrīda Circene
1.pielikums
Ministru kabineta
2013.gada 2.jūlija
noteikumiem Nr.354
Ziņojums kompetentajai iestādei par būtisku nevēlamu kosmētikas līdzekļa ietekmi




Veselības ministre Ingrīda Circene
Veselības ministrijas iesniegtajā redakcijā
2.pielikums
Ministru kabineta
2013.gada 2.jūlija
noteikumiem Nr.354
Kosmētikas līdzekļu paraugu ņemšana, paraugu sagatavošana un kosmētikas līdzekļu sastāvā esošo ķīmisko vielu noteikšanas metodes
1. Kosmētikas līdzekļu paraugu ņemšana
1. Kosmētikas līdzekļu kopējo paraugu (tādu atsevišķu paraugu kopums, kuriem ir viens un tas pats partijas numurs) ņem to oriģinālajos iepakojumos un neatvērtā veidā nosūta laboratorijai.
2. Kosmētikas līdzekļu, kas tirgū piedāvāti neiepakotā veidā vai mazumtirdzniecībā pārdoti tādā iepakojumā, kas atšķiras no ražotāja oriģinālā iepakojuma paraugus laboratorijas traukos (priekšmetos, kuros ievietots produkts un kuri ar to nepārtraukti saskaras) ņem tādā daudzumā, kāds nepieciešams analīžu veikšanai.
3. Laboratorijai nepieciešamo paraugu skaitu nosaka ar analītiskās metodes palīdzību.
4. Paraugus aizzīmogo (aizplombē) un etiķetes pievieno saskaņā ar normatīvajiem aktiem par kārtību, kādā tirgus uzraudzības iestādes pieprasa un saņem preču paraugus, kā arī rīkojas ar tiem pēc laboratoriskās vai cita veida ekspertīzes.
5. Paraugu ņemšanas un nodošanas aktu aizpilda saskaņā ar normatīvajiem aktiem par kārtību, kādā tirgus uzraudzības iestādes pieprasa un saņem preču paraugus, kā arī rīkojas ar tiem pēc laboratoriskās vai cita veida ekspertīzes.
6. Atsevišķie paraugi (vienības, kas ņemtas no pārdošanai piedāvātas partijas) jāglabā saskaņā ar ražotāja instrukcijām, kas norādītas uz marķējuma.
7. Laboratorijas paraugus (kopējā parauga reprezentatīva frakcija, kas jāanalizē atsevišķās laboratorijās) glabā tumšā vietā, 10–25 °C temperatūrā, ja vien nav precizēti citi nosacījumi.
8. Atsevišķos paraugus atver tieši pirms analīzes veikšanas.
2. Analizējamo daudzumu sagatavošana laboratorijā
9. Ja iespējams, katru atsevišķo paraugu analizē atsevišķi. Ja atsevišķais paraugs ir pārāk mazs, izmanto minimālo atsevišķo paraugu skaitu.
10. Pirms analizējamo daudzumu (reprezentatīvs laboratorijas parauga daudzums, kas nepieciešams vienai analīzei) ņemšanas atsevišķos paraugus labi sajauc.
11. Atbilstoši attiecīgajai analīzes metodei trauku, kurā glabā analizējamo paraugu, atver inertā gāzē un iespējami ātri vajadzīgā skaitā paņem analizējamos daudzumus. Analīzi izdara iespējami ātrāk. Ja paraugs jāsaglabā, inertā gāzē noslēdz trauku.
12. Ja sākotnēji homogēns kosmētikas līdzeklis sadalās pa fāzēm, pirms analizējamo daudzumu ņemšanas to vēlreiz homogenizē.
13. Ja kosmētikas līdzekli piedāvā tirgū veidā, kurā to nevar apstrādāt saskaņā ar šajā pielikumā noteiktajām metodēm, un ja attiecīgās analīžu metodes nav paredzētas šajā pielikumā, var pieņemt oriģinālu procedūru ar nosacījumu, ka to apraksta analīzes ziņojumā.
14. Šķidro kosmētikas līdzekļu (šķīdumi eļļā, spirtā vai ūdenī, tualetes ūdeņi, losjoni vai pieniņi, kuri var būt iepakoti flakonos, pudelēs, ampulās vai tūbiņās) analizējamo daudzumu ņem šādā kārtībā:
14.1. Trauku pirms atvēršanas spēcīgi sakrata.
14.2. Atver trauku.
14.3. Dažus mililitrus šķidruma ielej mēģenē, lai vizuāli novērtētu tā īpašības nolūkā paņemt analizējamo daudzumu.
14.4. Ja nepieciešams, paņem analizējamo daudzumu.
14.5. Rūpīgi noslēdz trauku.
15. Pusšķidru kosmētikas līdzekļu (pastas, krēmi, emulsijas un želejas, kuri var būt iepakoti tūbiņās, plastikas pudelēs vai burciņās) analizējamo daudzumu ņem šādā kārtībā:
15.1. No traukiem ar šauru kaklu noņem vismaz pirmo centimetru kosmētikas līdzekļa. Izspiež analizējamo daudzumu un tūlīt noslēdz trauku.
15.2. No traukiem ar platu kaklu vienmērīgi nokasa virsmu, atdalot virsējo slāni. Izņem analizējamo daudzumu un tūlīt noslēdz trauku.
16. Cietu kosmētikas līdzekļu (birstoši pūderi, presēti pūderi, zīmuļi, kuri var būt iepakoti dažādos traukos) analizējamo daudzumu ņem šādā kārtībā:
16.1. Birstošu pūderi pirms izpakošanas vai atvēršanas spēcīgi sakrata, atver un atdala analizējamo daudzumu.
16.2. No presēta pūdera vai zīmuļa vienmērīgi nokasa virsējo slāni. Analizējamo daudzumu ņem no atsegtā slāņa.
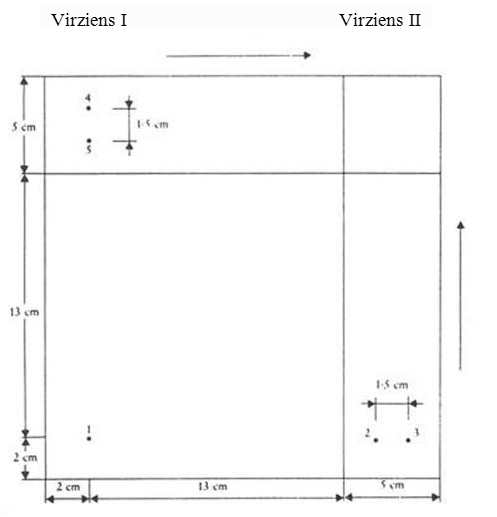
17. Aerosola (jebkura atkārtoti neizmantojama tvertne, kas izgatavota no metāla, stikla vai plastmasas un satur gāzi saspiestā, sašķidrinātā vai zem spiediena sašķidrinātā veidā ar vai bez šķidruma, pastas vai pulvera un ir apgādāta ar ierīci, kas ļauj saturam izplūst kā cietām vai šķidrām daļiņām kopā ar gāzi putu, pastas, pulvera vai šķidrā veidā) analizējamo daudzumu ņem šādi: aerosola trauku spēcīgi sakrata, reprezentatīvu tā satura daudzumu ar piemērotu savienotāju (piemēram, 1.attēls; izņemot gadījumus, kad analīzes metodē var paredzēt citus savienotājus) pārnes uz stikla pudeli, kas pārklāta ar plastiku (4.attēls) un kurai pielikts aerosola ventilis, bet kurai nav sifona caurules. Pārnesot daudzumu, pudeli tur ar ventili uz leju. Tā pārnesot, saturs kļūst skaidri redzams vienā no šādiem četriem veidiem:
17.1. Aerosols homogēna šķīduma veidā tiešai analīzei.
17.2. Aerosols, kas sastāv no divām šķidrām fāzēm. Katru fāzi var analizēt pēc tam, kad apakšējā fāze ir pārnesta uz citu pudeli, pirmo pudeli turot ar ventili uz leju. Apakšējā fāze bieži ir ūdeņaina, un tajā nav propelenta (piemēram, butāna un ūdens maisījuma).
17.3. Aerosols, kas satur suspendētu pulveri. Šķidro fāzi var analizēt pēc pulvera atdalīšanas.
17.4. Kosmētikas līdzeklis putu vai krēma veidā. Vispirms pudelē precīzi iesver 5–10 g 2-metoksietanola, tādējādi novēršot putu veidošanos degazācijas laikā un dodot iespēju izvadīt propelentās gāzes, nezaudējot šķidrumu.
18. Aerosola analizējamā daudzuma ņemšanā izmanto šādus piederumus:
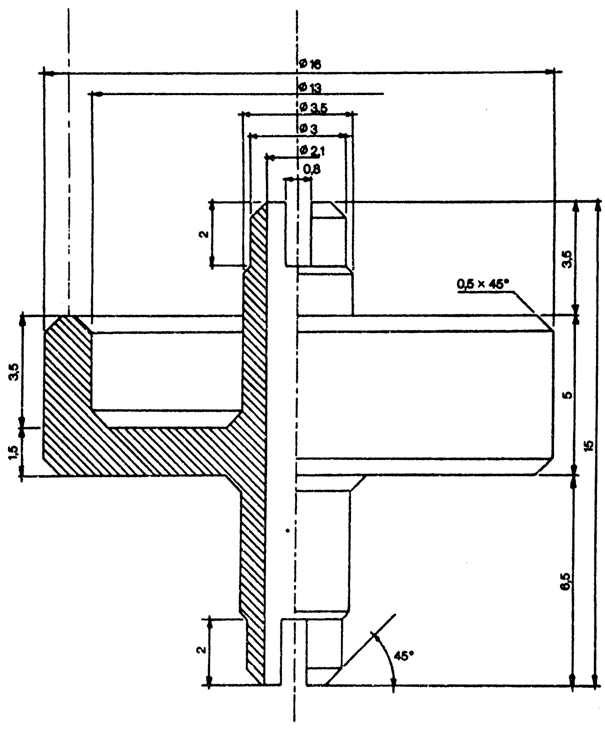
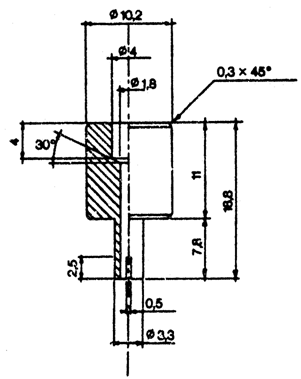
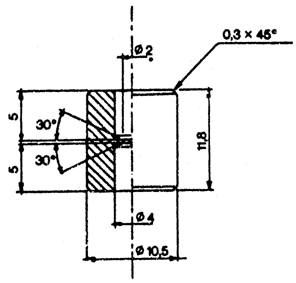
18.1. Savienotāju (1.attēls), kas izgatavots no dūralumīnija vai misiņa. Tas konstruēts tā, ka ar polietilēna adapteru to var pielāgot dažādām ventiļu sistēmām. Var lietot citus savienotājus (2. un 3.attēls).
18.2. Pudeli, kura aprakstīta šī pielikuma 17.punktā. Tās tilpums ir 50–100 ml.
19. Lai varētu pārnest pietiekamu daudzumu parauga, no pudeles izpūš gaisu. Šajā nolūkā pa savienotāju ievada aptuveni 10 ml dihlordifluormetāna vai butāna (atkarībā no analizējamā kosmētikas līdzekļa) un, turot pudeli ar ventili augstākajā stāvoklī, pilnīgi degazē, līdz izzūd šķidrā fāze. Noņem savienotāju. Nosver pudeli (a grami). Spēcīgi sakrata aerosolu, no kura ņemams paraugs. Pievieno savienotāju pie paraugam ņemamā aerosola trauka ventiļa (trauku turot ar ventili uz augšu), pieliek pudeli (ar kaklu uz leju) pie savienotāja. Piepilda apmēram divas trešdaļas pudeles. Ja pārnešana beidzas priekšlaicīgi tāpēc, ka izlīdzinājies spiediens, to var atjaunot, atdzesējot pudeli.
Šādi iegūto paraugu var izmantot:
19.1. Standarta ķīmiskā analīze:
19.1.1. Degazē, turot pudeli ar ventili uz augšu. Ja degazējot rodas putas, izmanto pudeli, kurā pa savienotāju ar šļirci iepriekš ievadīts precīzi nosvērts daudzums (5–10 g) 2-metoksietanola.
19.1.2. Pabeidz gaistošo komponentu atdalīšanu bez zudumiem, kratot ūdens vannā, kurā uztur 40 °C temperatūru. Noņem savienotāju.
19.1.3. Vēlreiz nosver pudeli (c grami), lai noteiktu atlikuma masu m2 (m2 = c – a) (aprēķinot atlikuma masu, atņem izmantotā 2-metoksietanola masu).
19.1.4. Noņemot ventili, atver pudeli.
19.1.5. Pilnīgi izšķīdina atlikumu zināmā daudzumā attiecīga šķīdinātāja.
19.1.6. Veic alikvotās daļas kvantitatīvu analīzi. Aprēķinos izmanto šādas formulas:
![]() un
un ![]() , kur
, kur
m1 – pudelē ievadītā aerosola masa;
m2 – atlikuma masa pēc karsēšanas 40 °C temperatūrā;
r – attiecīgās vielas procentuālais daudzums m2 (ko nosaka pēc attiecīgās metodes);
R – attiecīgās vielas procentuālais daudzums analīzei saņemtajā aerosolā;
Q – attiecīgās vielas kopējā masa aerosola traukā;
P – aerosola (atsevišķā parauga) tīrā sākuma masa.
19.2. Gaistošo komponentu analīze gāzu hromatogrāfijā:
19.2.1. Ar gāzu hromatogrāfijas šļirci no pudeles paņem attiecīgu daudzumu. Šļirces saturu ievada gāzu hromatogrāfā.
19.2.2. Izmanto A2 sērijas 25 µl vai 50 µ (5.attēls), vai līdzvērtīgu gāzu hromatogrāfijas šļirci precīzai parauga tilpuma nomērīšanai. Šai šļircei adatas galā ir pielikts ventilis. Ar pie pudeles piestiprinātu savienotāju un pie šļirces piestiprinātu polietilēna cauruli (garums 8 mm, iekšējais diametrs 2,5 mm) šļirce savienota ar pudeli.
19.2.3. Kad pudelē iepildīts attiecīgs daudzums aerosola, šļirces konisko galu pievieno pie pudeles atbilstoši šā pielikuma 19.2.2.apakšpunktam. Atver ventili un iesūknē atbilstīgu daudzumu šķidruma. Izvada gāzes burbuļus, vairākas reizes apgriežot virzuli (ja nepieciešams, atdzesē šļirci). Kad šļircē ir attiecīgais daudzums šķidruma bez burbuļiem, aizver ventili un atvieno šļirci no pudeles. Pieliek adatu, ievada šļirci gāzu hromatogrāfa inžektorā, atver ventili un ievada šļirces saturu.
19.2.4. Ja nepieciešams iekšējais standarts, to ievada pudelē (ar parastu stikla šļirci, izmantojot savienotāju).
1.attēls
Savienotājs P1
2.attēls
Savienotājs M2 pārejai starp aptveramajiem un
aptverošajiem ventiļiem
3.attēls
Savienotājs M1 pārejai starp diviem aptveramajiem
ventiļiem

4.attēls
Pudele. Ietilpība 50–100 ml
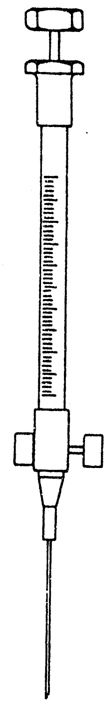
5.attēls
Šļirce gāzes paraugam
3. Brīvo nātrija un kālija hidroksīdu kvantitatīva un kvalitatīva noteikšana
20. Brīvo nātrija un kālija hidroksīdu kvantitatīvas un kvalitatīvas noteikšanas metode nosaka procedūru kvalitatīvai lielu daudzumu brīvo nātrija vai kālija hidroksīdu noteikšanai kosmētikas līdzekļos un kvantitatīvai brīvo nātrija vai kālija hidroksīdu noteikšanai matu ieveidošanas līdzekļos un šķīdinātājos, kas paredzēti nagu valnīša atbīdīšanai.
21. Brīvo nātrija un kālija hidroksīdu nosaka pēc skābes standartšķīduma tilpuma, kas nepieciešams, lai neitralizētu kosmētikas līdzekli, iegūto daudzumu izsakot brīvā nātrija hidroksīda masas %.
22. Laboratorijas paraugu izšķīdina vai disperģē ūdenī un titrē ar skābes standartšķīdumu. Pievienojot skābi, reģistrē pH vērtību: vienkārša nātrija vai kālija hidroksīda šķīduma beigu punkts ir straujš kāpums pH vērtībā. Vienkāršās titrēšanas līkni var izmainīt:
22.1. Amonjaks un citas vājas organiskās bāzes, kurām pašām titrēšanas līkne ir bez krasa lēciena. Pēc šīs metodes amonjaku atdala, destilējot vakuumā istabas temperatūrā.
22.2. Vāju skābju sāļi, kas titrēšanas līknē var radīt vairākus lēcienus. Tādā gadījumā tikai līknes sākumdaļa līdz pirmajam lēcienam atbilst brīvā nātrija vai kālija hidroksīda hidroksiljona neitralizācijai.
Ja vāju neorganisko skābju sāļi rada būtiski traucējošu ietekmi, veic alternatīvu titrēšanu spirtā. Teorētiski ir iespējams, ka augsto pH līmeni nosaka citas stipras šķīstošas bāzes (piemēram, litija hidroksīds, četraizvietotais amonija hidroksīds), tomēr to klātbūtnes varbūtība šī veida kosmētikas līdzekļos ir ļoti maza.
3.1. Kvalitatīva noteikšana
23. Reaģentam izmanto sārma standarta buferšķīdumu ar pH 9,18 25 °C temperatūrā: 0,05 M nātrija tetraborāta dekahidrāta.
24. Kvalitatīvai noteikšanai nepieciešama šāda iekārta:
24.1. Parastie laboratorijas stikla trauki.
24.2. pH metrs.
24.3. Stikla membrānas elektrods.
24.4. Kalomela standartelektrods.
25. pH metru kalibrē ar elektrodiem, izmantojot standarta buferšķīdumu. Sagatavo analizējamā kosmētikas līdzekļa 10 % šķīdumu vai dispersiju ūdenī un izfiltrē. Izmēra pH. Ja pH ir 12 vai vairāk, izdara kvantitatīvo noteikšanu.
3.2. Kvantitatīva noteikšana
26. Titrēšanu ūdens vidē veic šādā kārtībā:
26.1. Reaģentam izmanto standarta 0,1 n sālsskābi.
26.2. Titrēšanai ūdens vidē izmanto iekārtu:
26.2.1. Parastie laboratorijas stikla trauki.
26.2.2. pH metrs, vēlams ar pašrakstītāju.
26.2.3. Stikla membrānas elektrods.
26.2.4. Kalomela standartelektrods.
26.3. Vārglāzē ar 150 ml tilpumu precīzi iesver analizējamo daudzumu 0,5–1,0 g. Ja analizējamā daudzumā klāt ir amonjaks, pievieno dažus vārķermeņus, liek vārglāzi vakuumeksikatorā, pievieno ūdensstrūklas sūkni, līdz amonjaka smarža vairs nav konstatējama (apmēram trīs stundas).
26.4. Pievieno 100 ml ūdens, izšķīdina vai iemaisa atlikumu un, titrējot ar 0,1 n sālsskābes šķīdumu, reģistrē pH izmaiņas.
26.5. Nosaka kāpuma punktus titrēšanas līknēs. Ja pirmais kāpuma punkts ir zem pH 7 līmeņa, paraugā nav nātrija vai kālija hidroksīda.
26.6. Ja līknē ir divi vai vairāki kāpuma punkti, nozīmīgs ir tikai pirmais punkts.
26.7. Atzīmē titranta tilpumu pirmajā kāpuma punktā.
26.8. Titranta tilpumu apzīmē ar V, izsaka mililitros, analizējamā daudzuma masu gramos apzīmē ar M.
26.9. Nātrija vai kālija hidroksīdu saturu paraugā izsaka % un aprēķina pēc formulas:
![]() .
.
26.10. Ja pretēji norādēm par nātrija vai kālija hidroksīdu klātbūtni lielā daudzumā titrēšanas līknē neparādās skaidrs kāpuma punkts, kvantitatīvo noteikšanu atkārto izopropanolā.
27. Titrēšanu izopropanolā veic šādā kārtībā:
27.1. Titrēšanai izopropanolā izmantotie reaģenti ir šādi:
27.1.1. Izopropanols.
27.1.2. 1,0 n sālsskābes standartšķīdums.
27.1.3. Atšķaidot 1,0 n sālsskābi ar izopropanolu, tieši pirms lietošanas sagatavo 0,1 n sālsskābi izopropanolā.
27.2. Titrēšanai izopropanolā izmantotā iekārta:
27.2.1. Parastie laboratorijas stikla trauki.
27.2.2. pH metrs, vēlams ar pašrakstītāju.
27.2.3. Stikla membrānas elektrods.
27.2.4. Kalomela standartelektrods.
27.3. Vārglāzē ar 150 ml tilpumu precīzi iesver analizējamo daudzumu 0,5–1,0 g. Ja klāt ir amonjaks, pievieno dažus vārķermeņus, liek vārglāzi vakuumeksikatorā, pievieno ūdensstrūklas sūkni, līdz amonjaka smarža vairs nav konstatējama (apmēram trīs stundas). Pievieno 100 ml izopropanola, izšķīdina vai iemaisa atlikumu un, titrējot ar 0,1 n sālsskābes šķīdumu, reģistrē pH izmaiņas.
27.4. Aprēķinu veic saskaņā ar šī pielikuma 26.punktu, pieņemot, ka pirmais lēciena punkts ir apmēram pH 9 līmenī.
28. Ja nātrija vai kālija hidroksīda saturs nātrija hidroksīda izteiksmē ir 5 masas %, no viena parauga divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,25 % vērtību.4
4. Skābeņskābes un tās sārmu metālu sāļu kvantitatīva un kvalitatīva noteikšana matu kopšanas līdzekļos
29. Skābeņskābes un tās sārmu metālu sāļu kvantitatīvas un kvalitatīvas noteikšanas matu kopšanas līdzekļos metode nosaka procedūru skābeņskābes un tās sārmu metālu sāļu kvantitatīvai un kvalitatīvai noteikšanai matu kopšanas līdzekļos. To var izmantot bezkrāsainiem ūdens un spirta šķīdumiem un losjoniem, kas satur aptuveni 5 % skābeņskābes vai līdzvērtīgu daudzumu sārmu metālu oksalāta.
30. Ar šī pielikuma 29.punktā minēto metodi kvantitatīvi noteikto skābeņskābes vai tās sārmu metālu sāļu saturu izsaka parauga brīvās skābeņskābes masas procentos.
31. Pēc visu attiecīgo virsmas aktīvo anjonu aģentu atdalīšanas, kuri ir kopā ar p-toluidīna hidrohlorīdu, skābeņskābe vai oksalāti nogulsnējas kā kalcija oksalāts. Pēc tam šķīdumu izfiltrē, nogulsnes izšķīdina sērskābē un titrē ar kālija permanganātu.
32. Skābeņskābes un tās sārmu metālu sāļu kvantitatīvas un kvalitatīvas noteikšanas matu kopšanas līdzekļos metodē izmantojamie reaģenti1:
32.1. Amonija acetāta šķīdums, 5 masas %.
32.2. Kalcija hlorīda šķīdums, 10 masas %.
32.3. Etanols, 95 % (pēc tilpuma).
32.4. Tetrahlorogleklis.
32.5. Dietilēteris.
32.6. p-toluidīna dihidrohlorīda šķīdums, 6,8 masas %.
32.7. 0,1 N kālija permanganāta šķīdums.
32.8. Sērskābe, 20 masas %.
32.9. Sālsskābe, 10 masas %.
32.10. Nātrija acetāta trihidrāts.
32.11. Ledus etiķskābe.
32.12. Sērskābe (1:1).
32.13. Piesātināts bārija hidroksīda šķīdums.
33. Skābeņskābes un tās sārmu metālu sāļu kvantitatīvas un kvalitatīvas noteikšanas matu kopšanas līdzekļos metodē izmantojamās iekārtas:
33.1. Dalāmās piltuves, 500 ml.
33.2. Vārglāzes, 50 un 600 ml.
33.3. Stikla filtrtīģelis, G-4.
33.4. Mērcilindri, 25 un 100 ml.
33.5. Pipetes, 10 ml.
33.6. Bunzena kolbas, 500 ml.
33.7. Ūdensstrūklas sūknis.
33.8. Termometrs ar iedaļām no 0 līdz 100 °C.
33.9. Magnētiskais maisītājs ar sildelementu.
33.10. Magnētiskie stienīši ar teflona pārklājumu.
33.11. Birete, 25 ml.
33.12. Koniskās kolbas, 250 ml.
4.1. Kvantitatīva noteikšana
34. Iesver 6–7 g parauga 50 ml vārglāzē, ar atšķaidītu sālsskābi (atbilstoši šā pielikuma 32.9.apakšpunktam) noregulē pH uz 3 un ar 100 ml destilēta ūdens ieskalo dalāmajā piltuvē. Pēc kārtas pievieno 25 ml etanola, 25 ml p-toluidīna dihidrohlorīda šķīduma un 25–30 ml oglekļa tetrahlorīda un maisījumu spēcīgi sakrata.
35. Kad fāzes ir atdalītas, atdala apakšējo (organisko) fāzi, atkārto ekstrakciju ar šī pielikuma 34.punktā minētajiem reaģentiem un vēlreiz atdala organisko fāzi.
35. Ūdens šķīdumu ieskalo 600 ml vārglāzē un, vārot šķīdumu, atdala visu oglekļa tetrahlorīdu, kas nav atdalīts.
36. Pievieno 50 ml amonija acetāta šķīduma, karsē šķīdumu līdz viršanai un verdošajā šķīdumā iemaisa 10 ml karsta kalcija hlorīda šķīduma, ļauj nogulsnēties nogulsnēm.
37. Pievienojot dažus pilienus kalcija hlorīda šķīduma, pārbauda, vai nogulsnēšanās ir pilnīga, ļauj atdzist līdz istabas temperatūrai, tad iemaisa 200 ml etanola un atstāj uz 30 minūtēm.
38. Izfiltrē šķidrumu caur stikla filtrtīģeli, ar nelielu daudzumu karsta ūdens (50–60 °C) nogulsnes pārnes uz filtrtīģeli un mazgā ar aukstu ūdeni.
39. Nogulsnes piecas reizes mazgā ar mazu daudzumu etanola un pēc tam piecas reizes ar mazu daudzumu dietilētera, un izšķīdina nogulsnes 50 mililitros karstas sērskābes, sērskābi pazeminātā spiedienā izfiltrējot caur filtrtīģeli.
40. Šķīdumu bez zudumiem pārnes uz konisko kolbu un titrē ar kālija permanganāta šķīdumu, līdz šķīdums kļūst gaiši rozā.
41. Masas procentos izteiktu skābeņskābes saturu paraugā aprēķina pēc formulas:
% skābeņskābes = ![]() , kur
, kur
A – 0,1 N kālija permanganāta patēriņš, ko mēra saskaņā ar šī pielikuma 40.punktu;
E – analizējamā parauga daudzums (gramos) (34.punkts);
4,50179 – skābeņskābes pārvēršanas koeficients.
42. Ja skābeņskābes saturs ir aptuveni 5 %, no viena parauga divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,15 % vērtību.4
4.2. Kvalitatīva noteikšana
43. Skābeņskābe vai oksalāti nogulsnējas kā kalcija oksalāts un šķīst sērskābē. Šķīdumam pievieno mazliet kālija permanganāta šķīduma, kas kļūst bezkrāsains un izraisa oglekļa dioksīda veidošanos. Minēto oglekļa dioksīdu laižot caur bārija hidroksīda šķīdumu, veidojas baltas bārija karbonāta nogulsnes (duļķes).
44. Analizējamo parauga daudzumu apstrādā atbilstoši šā šī pielikuma 34., 35. un 36.punktam, tādējādi atdalot visus klātesošos mazgāšanas līdzekļus.
45. Aptuveni 10 mililitriem šķīduma, kas iegūts saskaņā ar šī pielikuma 44.punktu, pievieno lāpstiņas galu nātrija acetāta un šķīdumu skābina ar dažiem pilieniem ledus etiķskābes.
46. Pievieno 10 % kalcija hlorīda šķīduma un izfiltrē. Kalcija oksalāta nogulsnes izšķīdina 2 mililitros sērskābes (1:1).
47. Šķīdumu pārnes uz mēģeni un pa pilienam pievieno aptuveni 0,5 ml 0,1 N kālija permanganāta šķīduma. Ja šķīdumā ir oksalāts, šķīdums zaudē krāsu – sākumā pamazām, pēc tam strauji.
48. Tūlīt pēc tam, kad pievienots kālija permanganāts, mēģeni aiztaisa ar aizbāzni, kurā ir piemērota stikla caurule, mazliet pasilda mēģenes saturu un savāc izveidojušos oglekļa dioksīdu piesātinātā bārija hidroksīda šķīdumā. Ja pēc trim līdz piecām minūtēm redzamas baltas bārija karbonāta duļķes, šķīdumā ir skābeņskābe.
5. Hloroforma kvantitatīva noteikšana zobu pastā
49. Hloroforma kvantitatīvas noteikšanas zobu pastā metodi izmanto, lai gāzu hromatogrāfijā kvantitatīvi noteiktu hloroforma daudzumu zobu pastā. Šī metode ir piemērota hloroforma kvantitatīvai noteikšanai, ja tā līmenis ir 5 % vai mazāks. Kvantitatīvi noteikto hloroforma saturu izsaka kosmētikas līdzekļa masas procentos.
50. Zobu pastu suspendē dimetilformamīda un metanola maisījumā, kam pievieno zināmu daudzumu acetonitrila, kurš ir iekšējais standarts. Pēc centrifugēšanas daļu šķidrās fāzes ņem gāzu hromatogrāfijai un aprēķina hloroforma saturu.
51. Hloroforma kvantitatīvas noteikšanas zobu pastā metodei izmanto šādus reaģentus1:
51.1. Porapak Q, Chromosorb 101 vai līdzvērtīgs, kura daļiņu izmēri ir 80–100.
51.2. Acetonitrils.
51.3. Hloroforms.
51.4. Dimetilformamīds.
51.5. Metanols.
51.6. Iekšējais standartšķīdums, kuru pagatavo, ar pipeti 50 ml standarta kolbā iepilinot 5 ml dimetilformamīda un pievienojot aptuveni 300 mg (M mg) precīzi nosvērta acetonitrila. Uzpilda līdz atzīmei ar dimetilformamīdu un samaisa.
51.7. Šķīdumu, kas nepieciešams, lai noteiktu relatīvo signāla attiecību pret masas vienību, pagatavo, ar pipeti 10 ml standarta kolbā iepilinot 5 ml iekšējā standartšķīduma un pievienojot aptuveni 300 mg (M1 mg) precīzi svērta hloroforma. Uzpilda līdz zīmei ar dimetilformamīdu un samaisa.
52. Hloroforma kvantitatīvas noteikšanas zobu pastā metodei izmanto šādu iekārtu un aprīkojumu:
52.1. Analītiskie svari.
52.2. Gāzu hromatogrāfs ar liesmas jonizācijas detektoru.
52.3. Mikrošļirce ar ietilpību no 5–10 µl un 0,1 µl iedaļām.
52.4. Mērpipetes ar 1, 4 un 5 ml ietilpību.
52.5. Mērkolbas, 10 un 50 ml.
52.6. Mēģenes, apmēram 20 ml, ar skrūvējamiem vāciņiem, Sovirel France Nr.20 vai līdzvērtīgas. Skrūvējamā vāciņa iekšpusē ir blīvējoša plāksnīte, kas no vienas puses pārklāta ar teflonu.
52.7. Centrifūga.
53. Piemēroti apstākļi gāzu hromatogrāfijas veikšanai ir šādi:
53.1. Kolonnu (materiāls – stikls, garums 150 cm, iekšējais diametrs 4 mm, ārējais diametrs 6 mm) pilda, izmantojot vibratoru, arPorapak Q, Chromosorb 101 vai līdzvērtīgu materiālu, kura daļiņu izmēri ir 80–100.
53.2. Liesmu jonizācijas detektora jutību noregulē tā, lai, ievadot 3 µl šķīduma (atbilstoši šā pielikuma 51.7.apakšpunktam), acetonitrila pīķa augstums būtu aptuveni trīs ceturtdaļas no pilnas novirzes.
53.3. Nesējgāze – slāpeklis, plūsmas ātrums 65 ml/min. Gāzu pieplūdi detektoram noregulē tā, lai gaisa vai skābekļa plūsma 5–10 reizes pārsniegtu ūdeņraža plūsmu.
53.4. Temperatūra:
53.4.1. inžektorā 210 °C.
53.4.2. detektorā 210 °C.
53.4.3. kolonnas termostatā 175 °C.
53.5. Pašrakstītāja ātrums aptuveni 100 cm stundā.
54. Analizējamo paraugu ņem no neatvērtas tūbiņas. Noņem vienu trešo daļu satura, tūbiņai uzskrūvē atpakaļ vāciņu, rūpīgi sajauc tūbiņā un paņem analizējamo daudzumu.
55. Aizskrūvējamā mēģenē līdz tuvākajiem 10 mg iesver 6–7 g (Mo g) zobu pastas, kas apstrādāta saskaņā ar šī pielikuma 54.punktu, un pievieno trīs mazas stikla lodītes.
56. Ar pipeti mēģenē iepilina tieši 5 ml iekšējā standartšķīduma, 4 ml dimetilformamīda un 1 ml metanola, aizskrūvē mēģeni un sajauc.
57. Pusstundu krata ar mehānisko kratītāju un 15 minūtes aizskrūvēto mēģeni centrifugē tādā ātrumā, lai fāzes pilnīgi atdalītos. Ja šķidrā fāze paliek duļķaina arī pēc centrifugēšanas, to var mainīt, šķidrajai fāzei pievienojot 1–2 g nātrija hlorīda, ļaujot nogulsnēties un vēlreiz centrifugējot.
58. Atbilstoši šā pielikuma 53.punktam ievada 3 µl šā pielikuma 57.punktā minētā šķīduma. Atkārto minēto darbību. Atbilstoši iepriekš minētajiem nosacījumiem izdalīšanas laiku var izteikt ar šādām orientējošām vērtībām:
|
58.1. |
metanols |
apmēram viena minūte |
|
58.2. |
acetonitrils |
apmēram 2,5 minūtes |
|
58.3. |
hloroforms |
apmēram sešas minūtes |
|
58.4. |
dimetilformamīds |
>15 minūtes |
59. Lai noteiktu relatīvo signāla attiecību pret masas vienību, ievada 3 μl šķīduma (atbilstoši šā pielikuma 51.7.apakšpunktam). Atkārto šo darbību. Relatīvo signāla attiecību pret masas vienību nosaka katru dienu.
60. Relatīvā signāla attiecību pret masas vienību aprēķina šādi:
60.1. Izmēra acetonitrila un hloroforma pīķu augstumu un platumu pīķu pusaugstumā un aprēķina abu pīķu laukumu pēc formulas: augstums × platums pusaugstumā.
60.2. Nosaka acetonitrila un hloroforma pīķu laukumu hromatogrammās, kas iegūtas saskaņā ar šī pielikuma 59.punktu, un aprēķina relatīvo signāla attiecību fs pēc šādas formulas:
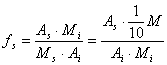 , kur
, kur
fs – hloroforma relatīvā signāla attiecība pret masas vienību;
As – hloroforma pīķa laukums (atbilstoši šā pielikuma 59.punktam);
A1 – acetonitrila pīķa laukums (atbilstoši šā pielikuma 59.punktam);
Ms – hloroforma daudzums (miligramos) 10 mililitros šķīduma, kas norādīts šā pielikuma 59.punktā (= M1);
Mi – acetonitrila daudzums (miligramos) 10 mililitros šķīduma, kas norādīts šā pielikuma 59.punktā (= 1/10 M).
Aprēķina iegūto rādījumu vidējo.
61. Hloroforma saturu aprēķina šādi:
61.1. Saskaņā ar šā pielikuma 60.1.apakšpunktu aprēķina hloroforma un acetonitrila pīķu laukumu hromatogrammās, kas iegūtas saskaņā ar šā pielikuma 58.punktā aprakstīto procedūru.
61.2. Hloroforma saturu zobu pastā aprēķina pēc šādas formulas:
![]() , kur
, kur
% X – hloroforma saturs zobu pastā, kas izteikts masas procentos;
As – hloroforma pīķa laukums (atbilstoši šā pielikuma 58.punktam);
Ai – acetonitrila pīķa laukums (atbilstoši šā pielikuma 58.punktam);
Msx – šā pielikuma 55.punktā norādītā parauga masa mg (= 1000 Mo);
Mi – acetonitrila daudzums (miligramos) 10 mililitros šķīduma, kas iegūts saskaņā ar šā pielikuma 56.punktu (1/10 M).
Aprēķina noteikto līmeņu vidējo un rezultātu izsaka ar 0,1 % precizitāti.
62. Ja hloroforma saturs ir aptuveni 3 %, no viena parauga divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,3 % vērtību.4
6. Cinka kvantitatīvā noteikšana
63. Cinka kvantitatīvās noteikšanas metodi piemēro hlorīda, sulfāta vai 4-hidroksibenzolsulfonāta, vai vairāku minēto cinka sāļu formā esoša cinka kvantitatīvajai noteikšanai kosmētikas līdzekļos.
64. Cinka saturu laboratorijas paraugā bis(2-metil-8-hinolīnoksīda) formā kvantitatīvi nosaka gravimetriski un izsaka paraugā esošās cinka masas procentos.
65. Šķīdumā esošais cinks skābā vidē nogulsnējas cinka bis(2-metil-8-hinolīnoksīda) formā. Pēc filtrēšanas nogulsnes izžāvē un nosver.
66. Cinka kvantitatīvās noteikšanas metodē izmanto šādus reaģentus1:
66.1. Koncentrēts amonjaks, 25 masas %; ![]() = 0,91.
= 0,91.
66.2. Ledus etiķskābe.
66.3. Amonija acetāts.
66.4. 2-metilhinolīn-8-ols.
66.5. Amonjaka šķīdums, 6 masas tilpuma %. Pārnes 240 g koncentrēta amonjaka uz 1000 ml mērkolbu, uzpilda līdz zīmei ar destilētu ūdeni un samaisa.
66.6. 0,2 M amonija acetāta šķīdums, ko iegūst, izšķīdinot 15,4 g amonija acetāta destilētā ūdenī, uzpilda līdz zīmei 1000 ml mērkolbā un samaisa.
66.7. 2-metilhinolīn-8-ola šķīdums, ko iegūst, izšķīdinot 5 g 2-metilhinolīn-8-ola 12 ml ledus etiķskābes un ar destilētu ūdeni pārnesot uz 100 ml mērkolbu. Uzpilda ar destilētu ūdeni līdz zīmei un samaisa.
67. Cinka kvantitatīvās noteikšanas metodē izmanto šādu iekārtu un aprīkojumu:
67.1. Mērkolbas, 100 un 1000 ml.
67.2. Vārglāzes, 400 ml.
67.3. Mērcilindri, 50 un 150 ml.
67.4. Mērpipetes, 10 ml.
67.5. Stikla filtrtīģeļi G-4.
67.6. Bunzena kolbas, 500 ml.
67.7. Ūdensstrūklas sūknis.
67.8. Termometrs ar iedaļām no 0 līdz 100 °C.
67.9. Eksikators ar piemērotu žāvētāju un mitruma indikatoru, piemēram, silikagelu vai līdzvērtīgu.
67.10. Žāvējamais skapis, kas noregulēts uz 150±2 °C temperatūru.
67.11. pH metrs.
67.12. Plītiņa.
68. Iesver 400 ml vārglāzē 5–10 g (turpmāk - M gramus) analizējamā parauga, kas satur apmēram 50–100 mg cinka, pievieno 50 ml destilēta ūdens un samaisa.
69. Uz katriem 10 mg cinka, kas ir šķīdumā (atbilstoši šā pielikuma 68.punktam), pievieno 2 ml 2-metilhinolīn-8-ola šķīduma un samaisa.
70. Šķīdumu atšķaida ar 150 ml destilēta ūdens, uzsilda līdz 60 °C un, nepārtraukti maisot, pievieno 45 ml 0,2 M amonija acetāta šķīduma.
71. Nepārtraukti maisot, ar 6 % amonjaka šķīdumu šķīduma pH noregulē intervālā no 5,7 līdz 5,9. Šķīduma pH mēra ar pH metru.
72. Šķīdumam 30 minūtes ļauj nostāties. Ar ūdensstrūklas sūkni izfiltrē caur G-4 filtrtīģeli, kas iepriekš izžāvēts (150 °C) un pēc atdzišanas nosvērts (turpmāk - M0 grami), un mazgā nogulsnes ar 150 ml destilēta ūdens 95 °C temperatūrā.
73. Tīģeli liek žāvējamā skapī 150 °C un žāvē vienu stundu.
74. Izņem tīģeli no žāvējamā skapja, liek eksikatorā un, kad tas ir atdzisis līdz istabas temperatūrai, nosaka tā masu (turpmāk - M1 grami).
75. Parauga cinka saturu masas procentos aprēķina pēc šādas formulas:
% cinka = ![]() , kur
, kur
M – atbilstoši šā pielikuma 68.punktam ņemtā parauga masa (gramos);
M0 – tukšā un sausā filtrtīģeļa (atbilstoši šā pielikuma 72.punktam) masa (gramos);
M1 – filtrtīģeļa un nogulšņu masa (gramos) (74.punkts).
76. No viena parauga ar aptuveni 1 masas % cinka saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,1 % vērtību.4
7. 4-hidroksibenzosulfoskābes kvantitatīva un kvalitatīva noteikšana
77. 4-hidroksibenzosulfoskābes kvantitatīvas un kvalitatīvas noteikšanas metode nosaka procedūru 4-hidroksibenzolsulfoskābes kvalitatīvai un kvantitatīvai noteikšanai aerosolos un sejas losjonos.
78. Kvantitatīvi noteikto 4-hidroksibenzolsulfoskābes saturu izsaka cinka 4-hidroksibenzolsulfonāta masas procentos kosmētikas līdzeklī.
79. Analizējamo daudzumu koncentrē pazeminātā spiedienā, šķīdina ūdenī un attīra, ekstrahējot ar hloroformu. Filtrēta ūdens šķīduma alikvotās daļas jodometrijā kvantitatīvi nosaka 4-hidroksibenzolsulfoskābi.
80. 4-hidroksibenzosulfoskābes kvantitatīvas un kvalitatīvas noteikšanas metodei izmanto šādus reaģentus1:
80.1. Koncentrēta sālsskābe, 36 masas % (![]() = 1,18).
= 1,18).
80.2. Hloroforms.
80.3. Butān-1-ols.
80.4. Ledus etiķskābe.
80.5. Kālija jodīds.
80.6. Kālija bromīds.
80.7. Nātrija karbonāts.
80.8. Sulfanilskābe.
80.9. Nātrija nitrāts.
80.10. 0,1 n kālija bromāts.
80.11. 0,1 n nātrija tiosulfāta šķīdums.
80.12. Cietes šķīdums ūdenī, 1 masas tilpuma %.
80.13. Nātrija karbonāta ūdens šķīdums, 2 masas tilpuma %.
80.14. Nātrija nitrīta ūdens šķīdums, 4,5 masas tilpuma %.
80.15. Ditizona šķīdums hloroformā, 0,05 masas tilpuma %.
80.16. Attīstīšanas šķīdinātājs: butān-1-ols/ledus etiķskābe/ūdens (tilpumu attiecībā 4:1:5). Samaisa dalāmajā piltuvē un izlej apakšējo fāzi.
80.17. Pauli reaģents, kuru iegūst šādi: karsējot izšķīdina 4,5 g sulfanilskābes, 45 ml koncentrētas sālsskābes un šķīdumu atšķaida ar ūdeni līdz 500 ml. Traukā ar ledus ūdeni atdzesē 10 ml šķīduma un maisot pievieno 10 ml auksta nātrija nitrīta šķīduma. Ļauj šķīdumam nostāties 15 minūtes 0 °C (šajā temperatūrā šķīdums ir stabils no vienas dienas līdz trijām dienām) un tieši pirms izsmidzināšanas (atbilstoši šā pielikuma 87.punktam) pievieno 20 ml nātrija karbonāta šķīduma.
80.18. Gatavas, ar celulozi pārklātas plāksnes (izmērs 20 × 20 cm, adsorbenta slāņa biezums 0,25 mm) plānslāņa hromatogrāfijai.
81. 4-hidroksibenzosulfoskābes kvantitatīvas un kvalitatīvas noteikšanas metodei izmanto šādas iekārtas un aprīkojumu:
81.1. Apaļkolbas ar slīpēta stikla aizbāzni, 100 ml.
81.2. Dalāmā piltuve, 100 ml.
81.3. Koniskā kolba ar slīpēta stikla aizbāzni, 250 ml.
81.4. Birete, 25 ml.
81.5. Mērpipetes, 1, 2 un 10 ml.
81.6. Mērpipete, 5 ml.
81.7. Mikrošļirce, 10 μl ar 0,1 μl iedaļām.
81.8. Termometrs ar iedaļām no 0 līdz 100 °C.
81.9. Ūdens vanna ar sildelementu.
81.10. Labi vēdināms žāvējamais skapis, kas noregulēts uz 80 °C temperatūru.
81.11. Standarta iekārta plānslāņa hromatogrāfijai.
82. Hidroksibenzolsulfoskābes kvalitatīvai un kvantitatīvai noteikšanai aerosolos izmanto atlikumu, ko iegūst, no aerosola trauka izlaižot šķīdinātājus un propelentus, kuri normālā spiedienā iztvaiko.
7.1. Kvalitatīva noteikšana
83. Ar mikrošļirci liek 5 µl atlikuma (atbilstoši šā pielikuma 82.punktam) vai parauga sešos punktos uz starta līnijas 1 cm no plānslāņa plāksnes apakšējās malas.
84. Liek plāksni attīstīšanas kamerā, kur jau ir attīstīšanas šķīdinātājs, un attīsta, līdz šķīdinātāja fronte ir 15 cm no starta līnijas.
85. Izņem plāksni no vannas un žāvē 80 °C, līdz vairs nav konstatējams etiķskābes tvaiks. Apsmidzina plāksni ar nātrija karbonāta šķīdumu un žāvē gaisā.
86. Pusi plāksnes pārsedz ar stiklu un nepārsegto daļu apsmidzina ar 0,05 % ditizona šķīdumu. Purpursarkanu plankumu veidošanās hromatogrammā liecina par cinka jonu klātbūtni.
87. Apsmidzināto plāksnes pusi pārsedz ar stiklu un otru pusi apsmidzina ar Pauli reaģentu. Hromatogrammā par 4-hidroksibenzolsulfoskābes klātbūtni liecina dzeltenbrūns plankums ar aptuvenu Rf vērtību 0,26, savukārt par 3-hidroksibenzolsulfoskābes klātbūtni liecina dzeltens plankums ar aptuvenu Rf vērtību 0,45.
7.2. Kvantitatīva noteikšana
88. Iesver 10 g parauga vai atlikuma (atbilstoši šā pielikuma 82.punktam) 100 ml apaļkolbā un ar rotācijas ietvaicētāju vakuumā ietvaicē gandrīz sausu virs ūdens vannas, kurā uztur 40 ºC.
89. Ar pipeti kolbā iepilina 10,0 ml (turpmāk -V1 ml) ūdens un karsējot izšķīdina ietvaicēšanas atlikumu (atbilstoši šā pielikuma 88.punktam).
90. Kvantitatīvi pārnes šķīdumu uz dalāmo piltuvi un divas reizes ekstrahē ūdens šķīdumu ar 20 ml hloroforma devu. Pēc katras ekstrakcijas izlej hloroforma fāzi.
91. Izfiltrē ūdens šķīdumu caur kroku filtru. Atkarībā no prognozējamā hidroksibenzolsulfoskābes satura ar pipeti iepilina 1,0 vai 2,0 ml (turpmāk - V2) filtrāta 50 ml koniskajā kolbā un atšķaida ar ūdeni līdz 75 ml.
92. Pievieno 2,5 ml 36 % sālsskābes un 2,5 g kālija bromīda, samaisa un ūdens vannā uzsilda līdz 50 ºC.
93. No biretes pievieno 0,1 n kālija bromāta, līdz šķīdums, kurš vēl ir 50 ºC temperatūrā, kļūst dzeltens.
94. Pievieno vēl 3,0 ml kālija bromāta šķīduma, aizbāž kolbu un ļauj 10 minūtes nostāties ūdens vannā. Ja pēc 10 minūtēm šķīdums zaudē krāsu, pievieno vēl 2,0 ml kālija bromāta šķīduma, aizbāž kolbu un 10 minūtes karsē virs ūdens vannas, kurā uztur 50 °C. Reģistrē kopējo pievienotā kālija bromāta šķīduma daudzumu (turpmāk - a).
95. Atdzesē šķīdumu līdz istabas temperatūrai, pievieno 2 g kālija jodīda un samaisa.
96. Radušos jodu titrē ar 0,1 n nātrija tiosulfāta šķīdumu. Titrēšanas beigu posmā pievieno dažus pilienus cietes šķīduma, kas ir indikators. Reģistrē izlietotā nātrija tiosulfāta daudzumu (turpmāk - b).
97. Aprēķina cinka hidroksibenzolsulfonāta saturu paraugā vai atlikumā (atbilstoši šā pielikuma 82.punktam) masas procentos pēc šādas formulas:
Masas % cinka hidroksibenzolsulfonāta = ![]() , kur
, kur
a – kopējais pievienotā 0,1 n kālija bromāta šķīduma (atbilstoši šā pielikuma 94.punktam) daudzums (mililitros);
b – attitrēšanai izlietotā 0,1 n nātrija tiosulfāta (atbilstoši šā pielikuma 96.punktam) daudzums (mililitros);
m – analizējamā kosmētikas līdzekļa vai atlikuma (atbilstoši šā pielikuma 88.punktam) daudzums (mililitros);
V1 – atbilstoši šā pielikuma 89.punktam iegūtā šķīduma tilpums (mililitros);
V2 – analīzei izlietotā izšķīdinātā ietvaicēšanas atlikuma (atbilstoši šā pielikuma 91.punktam) tilpums (mililitros).
98. Attiecībā uz aerosoliem mērījuma rezultātā jānorāda atlikums (atbilstoši šā pielikuma 82.punktam) masas % no sākotnējā kosmētikas līdzekļa daudzuma. Šajā aprēķinā pamatojas uz aerosolu paraugu ņemšanas noteikumiem.
99. No viena parauga, kas satur aptuveni 5 masas % cinka hidroksibenzolsulfonāta, divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,5 % vērtību.4
100. Rezultātu interpretācija: saskaņā ar Regulas (EK) Nr.1223/2009 3.pielikuma 25.punktu cinka 4-hidroksibenzolsulfonāta maksimālā pieļaujamā koncentrācija sejas losjonos un dezodorantos ir 6 masas %, un papildus hidroksibenzolsulfoskābes saturam nosaka arī cinka saturu. Reizinot aprēķināto cinka hidroksibenzolsulfonāta saturu (atbilstoši šā pielikuma 97.punktam) ar koeficientu 0,1588, iegūst minimālo cinka saturu masas %, kādam, ņemot vērā noteikto hidroksibenzolsulfoskābes saturu, teorētiski jābūt kosmētikas līdzeklī. Cinka saturs, ko faktiski nosaka gravimetriski, var būt augstāks, jo kosmētikas līdzekļos var izmantot arī cinka hlorīdu un cinka sulfātu.
8. Oksidējošo aģentu kvalitatīva noteikšana un ūdeņraža peroksīda kvantitatīva noteikšana matu kopšanas līdzekļos*
8.1. Persulfātu, bromātu un ūdeņraža peroksīda kvalitatīva noteikšana
101. Nātrija persulfātu, kālija persulfātu un amonija persulfātu, kālija bromātu, nātrija bromātu un ūdeņraža peroksīdu, kas ir vai nav radies no bārija peroksīda, kvalitatīvi nosaka papīra lejupejošajā hromatogrāfijā ar diviem attīstošajiem šķīdinātājiem.
102. Persulfātu, bromātu un ūdeņraža peroksīda kvalitatīvai noteikšanai izmanto šādus reaģentus1:
102.1. Šādu savienojumu 0,75 masas tilpuma % standartšķīdumi ūdenī:
102.1.1. Nātrija persulfāts.
102.1.2. Kālija persulfāts.
102.1.3. Amonija persulfāts.
102.1.4. Kālija bromāts.
102.1.5. Nātrija bromāts.
102.1.6. Ūdeņraža peroksīds.
102.2. Attīstošais šķīdinātājs A: 80 tilpuma % etanols.
102.3. Attīstošais šķīdinātājs B: benzols/metanols/3-metilbutān-1-ols/ūdens 34:38:18:10 (v/v/v/v).
102.4. Attīstošais reaģents A: 10 masas tilpuma % kālija jodīda šķīdums ūdenī.
102.5. Attīstošais reaģents B: 1 masas tilpuma % cietes šķīdums ūdenī.
102.6. Attīstošais reaģents C: 10 masas % sālsskābe.
102.7. 4 n sālsskābe.
103. Persulfātu, bromātu un ūdeņraža peroksīda kvalitatīvai noteikšanai izmanto šādu iekārtu un aprīkojumu:
103.1. Hromatogrāfijas papīrs (vatmaņpapīrs Nr.3 un Nr.4 vai līdzvērtīgs).
103.2. Mikropipete, 1 µl.
103.3. Mērkolbas, 100 ml.
103.4. Kroku filtri.
103.5. Iekārta papīra lejupejošajai hromatogrāfijai.
104. Ūdenī šķīstošo kosmētikas līdzekļu paraugu sagatavo, no katra parauga sagatavojot divus šķīdumus, izšķīdinot attiecīgi 1 g un 5 g kosmētikas līdzekļa 100 ml ūdens. Papīra hromatogrāfijā (atbilstoši šā pielikuma 107.punktam) izmanto 1 µl no katra šķīduma.
105. Ūdenī daļēji šķīstošo kosmētikas līdzekļu paraugu sagatavo šādi:
105.1. Nosver 1 g un 5 g parauga un disperģē 50 ml ūdens, uzpilda līdz 100 ml ar ūdeni un samaisa. Abas dispersijas izfiltrē caur kroku filtru un 1 µl no katra filtrāta izmanto papīra hromatogrāfijā (atbilstoši šā pielikuma 107.punktam).
105.2. Sagatavo vēl divas dispersijas no katra parauga, disperģējot 1 g un 5 g 50 ml ūdens, skābina ar atšķaidītu sālsskābi, uzpilda ar ūdeni līdz 100 ml un samaisa. Abas dispersijas izfiltrē caur kroku filtru, un 1 µl no katra filtrāta izmanto papīra hromatogrāfijā (atbilstoši šā pielikuma 107.punktam).
106. Krēmiem paraugu sagatavo, no katra kosmētikas līdzekļa 5 g un 20 g disperģējot 100 ml ūdens, un dispersijas izmanto papīra hromatogrāfijā (atbilstoši šā pielikuma 107.punktam).
107. Papīra hromatogrāfiju persulfātu, bromātu un ūdeņraža peroksīda kvalitatīvai noteikšanai veic šādā kārtībā:
107.1. Atbilstīgu daudzumu šķīdinātāja A (atbilstoši šā pielikuma 102.2.apakšpunktam) un B (atbilstoši šā pielikuma 102.3.apakšpunktam) pārnes uz divām atsevišķām hromatogrāfijas kamerām, lai veiktu papīra lejupejošo hromatogrāfiju. Hromatogrāfijas kameras vismaz 24 stundas piesātina ar šķīdinātāja tvaiku.
107.2. Katrā sākuma punktā uz hromatogrāfijas papīra (vatmaņpapīrs Nr.3 vai līdzvērtīgs) (atbilstoši šā pielikuma 103.1.apakšpunktam) joslas, kas ir 40 cm gara un 20 cm plata vai kurai ir cits piemērots lielums, liek 1µl viena parauga šķīduma un viena standartšķīduma, kas sagatavots atbilstoši šā pielikuma 104., 105., 106 un 102.1.apakšpunktam, un iztvaicē šķīdumu gaisā.
107.3. Hromatogrāfijas joslu (atbilstoši šā pielikuma 107.2.apakšpunktam) liek hromatogrāfijas kamerā, kas piepildīta ar attīstošo šķīdinātāju A (atbilstoši šā pielikuma 107.1.apakšpunktam), un attīsta, līdz šķīdinātāja fronte ir pavirzījusies par 35 cm (apmēram 15 stundas).
107.4. Atkārto šā pielikuma 107.2. un 107.3.apakšpunktā aprakstīto procedūru ar hromatogrāfijas papīru (vatmaņpapīru Nr.4 vai līdzvērtīgu) un attīstošo šķīdinātāju B. Veic hromatogrāfiju, līdz šķīdinātāja fronte pavirzījusies par 35 cm (apmēram piecas stundas).
107.5. Pēc attīstīšanas hromatogrammas izņem un žāvē gaisā.
107.6. Plankumus attīsta, hromatogrammu apsmidzinot pēc kārtas ar:
107.6.1. Attīstošo reaģentu A (atbilstoši šā pielikuma 102.4.apakšpunktam) un pēc īsa brīža ar attīstošo reaģentu B (atbilstoši šā pielikuma 102.5.apakšpunktam). Vispirms hromatogrammā parādās persulfātu plankumi, pēc tam ūdeņraža peroksīda plankumi. Plankumus atzīmē ar zīmuli.
107.6.2. Attīstošo reaģentu C (atbilstoši šā pielikuma 102.6.apakšpunktam), kas iegūts saskaņā ar šā pielikuma 107.6.1.apakšpunktu. Par bromātu klātbūtni liecina pelēcīgi zili plankumi hromatogrammā.
107.7. Ar iepriekšminētajiem nosacījumiem, kas attiecas uz attīstošajiem šķīdinātājiem A (atbilstoši šā pielikuma 102.2.apakšpunktam) un B (atbilstoši šā pielikuma 102.3.apakšpunktam), standartvielu (atbilstoši šā pielikuma 102.1.apakšpunktam) Rf vērtības ir aptuveni šādas:
|
Attīstošais šķīdinātājs A |
Attīstošais šķīdinātājs B |
|
|
Nātrija persulfāts |
0,40 |
0,10 |
|
Kālija persulfāts |
0,40 |
0,02 + 0,05 |
|
Amonija persulfāts |
0,50 |
0,10 + 0,20 |
|
Nātrija bromāts |
0,40 |
0,20 |
|
Kālija bromāts |
0,40 |
0,10 + 0,20 |
|
Ūdeņraža peroksīds |
0,80 |
0,80 |
8.2. Bārija peroksīda kvalitatīva noteikšana
108. Kad paraugs (atbilstoši šā pielikuma 105.punktam) skābināts, pēc ūdeņraža peroksīda veidošanās un bārija jonu klātbūtnes kvalitatīvi nosaka bārija peroksīdu šādā kārtībā:
108.1. Ja persulfātu nav (atbilstoši šā pielikuma 8.1.apakšnodaļai), tad, pievienojot atšķaidītu sērskābi skābā parauga šķīduma devai (atbilstoši šā pielikuma 111.1.apakšpunktam), veidojas baltas bārija sulfāta nogulsnes. Bārija jonu klātbūtni paraugā (atbilstoši šā pielikuma 111.1.apakšpunktam) atkal apstiprina papīra hromatogrāfijā saskaņā ar šā pielikuma 112.punktu.
108.2. Ja ir gan bārija peroksīds, gan persulfāti (atbilstoši šā pielikuma 111.2.apakšpunktam), tad šķīduma atlikumu (atbilstoši šā pielikuma 111.2.apakšpunktam) šķeļ sārmā. Pēc šķīdināšanas sālsskābē bārija jonu klātbūtni kausējuma šķīdumā (atbilstoši šā pielikuma 111.2.3.apakšpunktam) apstiprina papīra hromatogrāfijā un (vai) izgulsnējot bārija sulfāta veidā.
109. Bārija peroksīda kvalitatīvai noteikšanai izmanto šādus reaģentus1:
109.1. Metanols.
109.2. Koncentrēta sālsskābe, 36 % (m/m).
109.3. 6 n sālsskābe.
109.4. 4 n sērskābe.
109.5. Dinātrija rodizonāts
109.6. Bārija hlorīds (BaCl2·2H2O).
109.7. Bezūdens nātrija karbonāts.
109.8. Bārija hlorīda 1 % šķīdums ūdenī.
109.9. Attīstošais šķīdinātājs: metanols/koncentrēta (36 %) sālsskābe/ūdens 80:10:10 (v/v/v).
109.10. Attīstošais reaģents: 0,71 % (m/v) dinātrija rodizonāta šķīdums, ko sagatavo tieši pirms izmantošanas.
110. Bārija peroksīda kvalitatīvai noteikšanai izmanto šādu iekārtu un aprīkojumu:
110.1. Mikropipete, 5µl.
110.2. Platīna tīģeļi.
110.3. Mērkolbas, 100 ml.
110.4. Hromatogrāfijas papīrs (Schleicher un Schüll 2043 b vai līdzvērtīgs). Papīru notīra, attīsta hromatogrāfijas kamerā (atbilstoši šā pielikuma 103.5.apakšpunktam), kurā ir attīstošais šķīdinātājs (atbilstoši šā pielikuma 109.9.apakšpunktam), un nožāvē.
110.5. Kroku filtrpapīrs.
110.6. Parastā iekārta papīra augšupejošajai hromatogrāfijai.
111. Paraugu bārija peroksīda kvalitatīvai noteikšanai sagatavo šādi:
111.1. Kosmētikas līdzekļiem, kuros nav persulfātu:
111.1.1. Disperģē 2 g kosmētikas līdzekļa 50 ml ūdens un ar sālsskābi (atbilstoši šā pielikuma 109.3.apakšpunktam) noregulē dispersijas pH apmēram uz 1.
111.1.2. Pārnes dispersiju uz 100 ml mērkolbu, uzpilda līdz zīmei ar ūdeni un samaisa. Dispersiju izmanto papīra hromatogrāfijas analīzei (atbilstoši šā pielikuma 112.punktam) un bārija kvalitatīvai noteikšanai, izgulsnējot sulfāta veidā.
111.2. Kosmētikas līdzekļiem, kuros ir persulfāti:
111.2.1. Disperģē 2 g kosmētikas līdzekļa 100 ml ūdens un izfiltrē.
111.2.2. Izžāvētajam atlikumam pievieno nātrija karbonātu (atbilstoši šā pielikuma 109.7.apakšpunktam), kura masa ir 7–10 reizes mazāka par minētā atlikuma masu, sajauc un maisījumu pusstundu kausē platīna tīģelī.
111.2.3. Atdzesē līdz istabas temperatūrai, izšķīdina kausējumu 50 ml ūdens un izfiltrē (atbilstoši šā pielikuma 110.5.apakšpunktam).
111.2.4. Kausējuma atlikumu izšķīdina sālsskābē (atbilstoši šā pielikuma 109.3.apakšpunktam) un uzpilda ar ūdeni līdz 100 ml. Šķīdumu izmanto papīra hromatogrāfijas analīzei (atbilstoši šā pielikuma 112.punktam) un bārija kvalitatīvai noteikšanai, izgulsnējot sulfāta veidā.
112. Bārija peroksīda kvalitatīvo noteikšanu veic šādā kārtībā:
112.1. Attiecīgu daudzumu attīstošā šķīdinātāja (atbilstoši šā pielikuma 109.9.apakšpunktam) pārnes uz augšupejošās papīra hromatogrāfijas kameru un piesātina kameru vismaz 15 stundas.
112.2. Uz hromatogrāfijas papīra gabala, kas iepriekš apstrādāts saskaņā ar šā pielikuma 110.4.apakšpunktu, trijos sākuma punktos liek 5 µl katra šķīduma, kas sagatavots saskaņā ar šā pielikuma 111.1.2. un 111.2.4.apakšpunktu, un standartšķīduma (atbilstoši šā pielikuma 109.8.apakšpunktam).
112.3. Gaisā nožāvē parauga un standartšķīduma plankumus. Attīsta hromatogrammu, līdz šķīdinātāja fronte ir pavirzījusies par 30 cm.
112.4. Hromatogrammu izņem no kameras un žāvē gaisā.
112.5. Apsmidzinot papīru ar attīstošo reaģentu (atbilstoši šā pielikuma 109.10.apakšpunktam), attīsta plankumus hromatogrammā. Bārija klātbūtnē hromatogrammā parādās sarkani plankumi ar aptuvenu Rf vērtību 0,10.
8.3. Ūdeņraža peroksīda kvantitatīva noteikšana
113. Ūdeņraža peroksīda jodometriskās kvantitatīvās noteikšanas pamatā ir šāda reakcija:
H2O2 + 2H+ + 2I– ![]() I2 +
2H2O.
I2 +
2H2O.
Reakcija notiek lēni, bet to var paātrināt, pievienojot amonija molibdātu. Radušos jodu nosaka titrimetriski ar nātrija tiosulfātu, un tas ir ūdeņraža peroksīda satura mērs.
114. Ūdeņraža peroksīda saturu mēra saskaņā ar šā pielikuma 115., 116., 117., 118. un 119.punktu un izsaka kosmētikas līdzekļa masas procentos (% m/m).
115. Ūdeņraža peroksīda kvantitatīvai noteikšanai izmanto šādus reaģentus1:
115.1. 2 n sērskābe.
115.2. Kālija jodīds.
115.3. Amonija molibdāts.
115.4. 0,1 n nātrija tiosulfāts.
115.5. 10 % (m/v) kālija jodīda šķīdums, ko gatavo tieši pirms izmantošanas.
115.6. Amonija molibdāta šķīdums, 20 % (m/v).
115.7. Cietes šķīdums, 1 % (m/v).
116. Ūdeņraža peroksīda kvantitatīvai noteikšanai izmanto šādu iekārtu un aprīkojumu:
116.1. Vārglāzes, 100 ml.
116.2. Birete, 50 ml.
116.3. Mērkolbas, 250 ml.
116.4. Mērcilindri, 25 un 100 ml.
116.5. Pipetes ar vienu iedaļu, 10 ml.
116.6. Koniskās kolbas, 250 ml.
117. Ūdeņraža peroksīda kvantitatīvo noteikšanu veic šādā kārtībā:
117.1. Vārglāzē ar 100 ml ietilpību iesver 10 g (turpmāk - m gramus) kosmētikas līdzekļa, kas satur 0,6 g ūdeņraža peroksīda. Pārnes vārglāzes saturu uz 250 ml mērkolbu, uzpilda ar ūdeni līdz atzīmei un samaisa.
117.2. Ar pipeti iepilina 10 ml parauga šķīduma (atbilstoši šā pielikuma 117.1.apakšpunktam) 250 ml koniskajā kolbā (atbilstoši šā pielikuma 116.6.apakšpunktam) un secīgi pievieno 100 ml 2 n sērskābes (atbilstoši šā pielikuma 115.1.apakšpunktam), 20 ml kālija jodīda šķīduma (atbilstoši šā pielikuma 115.5.apakšpunktam) un trīs pilienus amonija molibdāta šķīduma (atbilstoši šā pielikuma 115.6.apakšpunktam).
117.3. Jodu, kas veidojas, tūlīt titrē ar 0,1 n nātrija tiosulfāta šķīdumu (atbilstoši šā pielikuma 115.4.apakšpunktam) un tieši pirms beigu punkta kā indikatoru pievieno dažus mililitrus cietes šķīduma (atbilstoši šā pielikuma 115.7.apakšpunktam). Reģistrē 0,1 n nātrija tiosulfāta (atbilstoši šā pielikuma 115.4.apakšpunktam) patēriņu mililitros (turpmāk - V).
117.4. Saskaņā ar šā pielikuma 117.2. un 117.3.apakšpunktu izdara tukšo analīzi, 10 ml parauga šķīduma vietā izmantojot 10 ml ūdens. Reģistrē 0,1 n nātrija tiosulfāta patēriņu tukšajā analīzē (turpmāk - Vo ml).
118. Aprēķina ūdeņraža peroksīda saturu kosmētikas līdzeklī masas procentos (% m/m) pēc formulas:
% ūdeņraža peroksīda = (((V – Vo) × 1,7008 × 250 × 100)/(m × 10 × 1000))
vai
% ūdeņraža peroksīda = (((V – Vo) × 4,252)/(m)), kur
m – analizējamā kosmētikas līdzekļa (atbilstoši šā pielikuma 117.1.apakšpunktam) daudzums (gramos);
Vo – 0,1 n tiosulfāta šķīduma patēriņš (mililitros) tukšajā analīzē (atbilstoši šā pielikuma 117.4.apakšpunktam);
V – 0,1 n tiosulfāta šķīduma patēriņš (mililitros) parauga šķīduma titrēšanā (atbilstoši šā pielikuma 117.3.apakšpunktam).
119. Ja ūdeņraža peroksīda saturs kosmētikas līdzeklī ir aptuveni 6 %, no viena parauga divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,2 % vērtību.4
* Piezīme. Ūdeņraža peroksīdu kosmētikas līdzekļos jodometriski var noteikt tikai tad, ja tajos nav citu oksidējošu aģentu, kas no jodīdiem veido jodu. Tāpēc pirms ūdeņraža peroksīdu kvantitatīvas noteikšanas jodometriski, kvalitatīvi un kvantitatīvi nosaka visus pārējos kosmētikas līdzeklī esošos oksidējošos aģentus. Kvalitatīva noteikšana notiek divos posmos – pirmajā nosaka persulfātus, bromātus un ūdeņraža peroksīdu un otrajā nosaka bārija peroksīdu.
9. Dažu oksidējošo krāsvielu kvalitatīva un puskvantitatīva noteikšana matu krāsās
120. Oksidējošo krāsvielu kvalitatīva un puskvantitatīva noteikšana matu krāsās ir piemērota šādu vielu kvalitatīvai un puskvantitatīvai noteikšanai krēmveida vai šķidrās matu krāsās:
|
Vielas |
Simbols |
|
fenilēndiamīni |
|
|
o-fenilēndiamīns |
(OPD) |
|
m-fenilēndiamīns |
(MPD) |
|
p-fenilēndiamīns |
(PPD) |
|
metilfenilēndiamīni |
|
|
4-metil-1,2-fenilēndiamīns (toluol-3,4-diamīns) |
(OTD) |
|
4-metil-1,3-fenilēndiamīns (toluol-2,4-diamīns) |
(MTD) |
|
2-metil-1,4-fenilēndiamīns (toluol-2,5-diamīns) |
(PTD) |
|
diaminofenoli |
|
|
2,4-diaminofenols |
(DAP) |
|
hidrohinons |
|
|
1,4-benzēndiols |
(H) |
|
α-naftols |
(a-N) |
|
pirogallols |
|
|
1,2,3-hidroksibenzols |
(P) |
|
rezorcīns |
|
|
1,3-dihidroksibenzols |
(R) |
121. Oksidējošās krāsvielas pH 10 līmenī ar 96 % etanolu ekstrahē no krēmveida vai šķidrām matu krāsām un kvalitatīvi nosaka viendimensionālā vai divdimensionālā plānslāņa hromatogrāfijā. Veicot šo vielu puskvantitatīvu noteikšanu, paraugu hromatogrammu četrās attīstīšanas sistēmās salīdzina ar vienlaicīgi un iespējami līdzīgos apstākļos iegūtu standartvielu hromatogrammu.
122. Oksidējošo krāsvielu kvalitatīvai un puskvantitatīvai noteikšanai matu krāsās izmanto šādus reaģentus1:
122.1. Etanols, bezūdens.
122.2. Acetons.
122.3. Etanols, 96 % (v/v).
122.4. Amonjaka šķīdums, 25 % (d204 = 0,91).
122.5. L(+)-askorbīnskābe.
122.6. Hloroforms.
122.7. Cikloheksāns.
122.8. Slāpeklis, tehniskais.
122.9. Toluols.
122.10. Benzols.
122.11. n-butanols.
122.12. 2-butanols.
122.13. Fosforpaskābe, 50 % (v/v) šķīdums.
122.14. Diazotējošs reaģents: 3-nitro-1-benzoldiazonija hlorbenzolsulfonāts (stabilizēta sāls forma) kā Red 2 JN–Francolor vai 2-hlor-4-nitro-1-benzoldiazonija naftalīnbenzoāts (stabilizēta sāls forma) kā NNCD reaģents Nr.74 150 FLUKA, vai līdzvērtīgs.
122.15. Sudraba nitrāts.
122.16. p-dimetilaminobenzaldehīds.
122.17. 2,5-dimetilfenols.
122.18. Dzelzs(III) hlorīda heksahidrāts.
122.19. Sālsskābe, 10 % (m/v) šķīdums.
122.20. Standartvielas, kuras minētas šī pielikuma 120.punktā. Amīnu savienojumu standartviela ir hidrohlorīdam (mono- vai di-) vai brīvai bāzei.
122.21. Standartšķīdumi 0,5 % (m/v):
122.21.1. Sagatavo visu šī pielikuma 122.20.apakšpunktā minēto standartvielu 0,5 % (m/v) šķīdumus.
122.21.2. Iesver 50±1 mg standartvielas 10 ml mērkolbā.
122.21.3. Pievieno 5 ml 96 % etanola (atbilstoši šā pielikuma 122.3. apakšpunktam) un 250 mg askorbīnskābes (atbilstoši šā pielikuma 122.5. apakšpunktam).
122.21.4. Lai šķīdums būtu sārmains, pievieno amonjaka šķīdumu (atbilstoši šā pielikuma 122.4.apakšpunktam), kas nodrošina pH 10 (pārbauda ar indikatorpapīru).
122.21.5. Uzpilda līdz 10 ml ar 96 % etanolu (atbilstoši šā pielikuma 122.3.apakšpunktam) un samaisa.
122.21.6. Šķīdumus var glabāt vienu nedēļu tumšā, vēsā vietā.
122.21.7. Dažos gadījumos pēc askorbīnskābes un amonjaka pievienošanas var veidoties nogulsnes. Pirms turpina analīzi, nogulsnēm ļauj nostāties.
122.22. Attīstošie šķīdinātāji. Attīstošie šķīdinātāji, kas satur amonjaku, tieši pirms izmantošanas labi jāsakrata:
122.22.1. Acetons/hloroforms/toluols 35:25:40 (v/v/v).
122.22.2. Hloroforms/cikloheksāns/absolūtais spirts/25 % amonjaks 80:10:10:1 (v/v/v/v).
122.22.3. Benzols/butān-2-ols/ūdens 50:25:25 (v/v/v). Labi sakrata un pēc atdalīšanas istabas temperatūrā (20–25 °C) izmanto virsējo fāzi.
122.22.4. n-butanols/hloroforms/reaģents M 7:70:23 (v/v/v). Istabas temperatūrā (20–25 °C) rūpīgi atdala un izmanto apakšējo fāzi.
|
Reaģenta M gatavošana |
|
|
Amonjaka šķīdums, 25 % (v/v) |
24 tilp.v. |
|
Fosforpaskābe, 50 % (122.13.apakšpunkts) |
1 tilp.v. |
|
Ūdens |
75 tilp.v. |
122.23. Attīstītāji:
122.23.1. Diazotējošs reaģents. Sagatavo attiecīgā reaģenta 5 % (m/v) šķīdumu ūdenī (atbilstoši šā pielikuma 122.14.apakšpunktam). Šķīdums jāgatavo tieši pirms izmantošanas.
122.23.2. Ērliha reaģents. Izšķīdina 2 g p-dimetilaminobenzaldehīda (atbilstoši šā pielikuma 122.16.apakšpunktam) 100 ml 10 % (m/v) sālsskābes šķīduma ūdenī (atbilstoši šā pielikuma 122.19.apakšpunktam).
122.23.3. 2,5-dimetilfenols – dzelzs(III) hlorīda heksahidrāts:
Pirmais šķīdums: izšķīdina 1 g dimetilfenola (atbilstoši šā pielikuma 122.17.apakšpunktam) 100 ml 96 % etanola (atbilstoši šā pielikuma 122.3.apakšpunktam).
Otrais šķīdums: izšķīdina 4 g dzelzs(III) hlorīda heksahidrāta (atbilstoši šā pielikuma 122.18.apakšpunktam) 100 ml 96 % etanola (atbilstoši šā pielikuma 122.3.apakšpunktam).
Attīstot šos divus šķīdumus izsmidzina atsevišķi – vispirms pirmo šķīdumu, pēc tam otro šķīdumu.
122.23.4. Sudraba nitrāta amonjakāls šķīdums. 5 % (m/v) sudraba nitrāta šķīdumam ūdenī (atbilstoši šā pielikuma 122.15.apakšpunktam) pievieno 25 % amonjaku (atbilstoši šā pielikuma 122.4.apakšpunktam), līdz nogulsnes izšķīst. Šis reaģents jāsagatavo tieši pirms izmantošanas. Neglabā.
123. Oksidējošo krāsvielu kvalitatīvai un puskvantitatīvai noteikšanai matu krāsās izmanto šādu iekārtu:
123.1. Parastais laboratorijas aprīkojums plānslāņa hromatogrāfijai:
123.1.1. Plastikas vai stikla pārsegs, kas izveidots tā, lai hromatogrāfiskā plate, uznesot šķīdumus un žāvējot, atrastos slāpekļa atmosfērā. Šī piesardzība vajadzīga tāpēc, lai novērstu to, ka dažas krāsvielas oksidējas.
123.1.2. Mikrošļirce, 10 µl, ar 0,2 µl iedaļām un adatu ar taisnu galu vai – labāk – 50µl automātiskais dozators, ko nostiprina statīvā tā, lai plati varētu turēt slāpeklī.
123.1.3. Izmantošanai gatavas 0,25 mm biezas 20 × 20 cm plānslāņu silikagela plates (Macherey and Nagel, Silica G-HR, kam ir plastikas pamats, vai līdzvērtīgas).
123.2. Centrifūga, 4000 apgr./min.
123.3. Centrifūgas mēģenes, 10 ml, ar skrūvējamu plastikas vāciņu, kas pārklāts ar PTFE, vai līdzvērtīgas.
124. Oksidējošo krāsvielu kvalitatīvu un puskvantitatīvu noteikšanu matu krāsās veic šādā kārtībā:
124.1. Analizējamo paraugu apstrādi veic šādi:
124.1.1. Noņem pirmos 2 vai 3 cm krēma, ko izspiež no tūbiņas.
124.1.2. Iepriekš ar slāpekli izskalotā centrifūgas mēģenē (atbilstoši šā pielikuma 123.3.apakšpunktam) liek 300 mg askorbīnskābes un 3 g krēma vai 3 g homogenizēta šķidruma.
124.1.3. Pa pilienam pievieno 25 % amonjaku, līdz pH ir 10.
124.1.4. Uzpilda līdz 10 ml ar 96 % etanolu (atbilstoši šā pielikuma 122.3.apakšpunktam).
124.1.5. Homogenizē slāpeklī (atbilstoši šā pielikuma 122.8.apakšpunktam), aiztaisa mēģeni un 10 minūtes centrifugē ar 4000 apgr./min.
124.1.6. Izmanto centrifugātu.
124.2. Hromatogrāfija:
124.2.1. Šķīdumu uznešana uz platēm:
124.2.1.1. Slāpeklī (atbilstoši šā pielikuma 122.8.apakšpunktam) uz hromatogrāfijas plates (atbilstoši šā pielikuma 123.1.3.apakšpunktam) deviņos punktos, kas atrodas apmēram 1,5 cm cits no cita uz līnijas, kura ir apmēram 1,5 cm no plates malas, klāj 1 µl katra iepriekšminētā standartšķīduma.
124.2.1.2. Standartšķīduma plankumus izvieto šādi:
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
R |
P |
H |
PPD |
DAP |
PTD |
OPD |
OTD |
MPD |
|
MTD |
a-N |
124.2.1.3. Aiz devītā standartšķīduma plankuma (MPD) klāj attiecīgi 2 µl kontrolšķīduma paraugu, ko iegūst saskaņā ar šī pielikuma 124.1.apakšpunktu.
124.2.1.4. Plati tur slāpeklī (atbilstoši šā pielikuma 122.8.apakšpunktam) līdz hromatogrāfiskās analīzes beigām.
124.2.2. Attīstīšana:
124.2.2.1. Liek plati kamerā, kas iepriekš izskalota ar slāpekli (atbilstoši šā pielikuma 122.8.apakšpunktam), piesātina ar vienu no četriem šķīdinātājiem (atbilstoši šā pielikuma 122.22.apakšpunktam) un attīsta istabas temperatūrā (20–25 °C) tumsā, līdz šķīdinātāja fronte ir pavirzījusies apmēram par 15 cm no starta līnijas.
124.2.2.2. Izņem plati un istabas temperatūrā žāvē slāpeklī (atbilstoši šā pielikuma 122.8.apakšpunktam).
124.2.3. Plati tūlīt apsmidzina ar vienu no četriem šķīdinātājiem, kas norādīti šā pielikuma 122.23.apakšpunktā.
124.2.4. Kvalitatīva noteikšana. Salīdzina parauga Rf vērtību un iegūto krāsu ar hromatografēto standartvielu Rf vērtību un krāsu. Šā pielikuma I tabulā ir Rf vērtību piemēri un krāsas katrai standartvielai atkarībā no izmantotā šķīdinātāja un indikatora. Šaubu gadījumā apstiprinājumu dažreiz var gūt ar standartpievienošanas metodi, pievienojot attiecīgās standartvielas šķīdumu parauga ekstraktam.
124.2.5. Puskvantitatīvais vērtējums. Vizuāli salīdzina visu šā pielikuma 124.2.4.apakšpunktā noteikto vielu plankumu intensitāti ar standartvielu attiecīgā diapazona koncentrācijām. Ja vienas vai vairāku paraugā atrasto vielu koncentrācija ir pārākumā, parauga ekstraktu atšķaida un mērījumu atkārto.
I tabula
Rf vērtības un krāsas, ko iegūst tūlīt pēc attīstīšanas
|
Standartviela (122.20.) |
Attīstošie šķīdinātāji |
Attīstītāji |
||||||
|
Rf vērtības |
iegūtās krāsas |
|||||||
|
(122.22.1.) |
(122.22.2.) |
(122.22.3.) |
(122.22.4.) |
diazotējošs reaģents (122.23.1.) |
Ērliha reaģents (122.23.2.) |
dimetilfenols (122.23.3.) |
AgNO3(122.23.4.) |
|
|
OPD |
0,62 |
0,60 |
0,30 |
0,57 |
bāli brūns |
– |
– |
bāli brūns |
|
MPD |
0,40 |
0,60 |
0,47 |
0,48 |
violeti brūns* |
dzeltens |
bāli brūns |
bāli brūns |
|
PPD |
0,20 |
0,50 |
0,30 |
0,48 |
brūns |
spilgti sarkans* |
violets |
pelēks |
|
OTD |
0,60 |
0,60 |
0,53 |
0,60 |
brūns* |
bāli oranžs |
bāli brūns |
pelēcīgi brūns |
|
MTD |
0,40 |
0,67 |
0,45 |
0,60 |
sarkanīgi brūns* |
dzeltens |
brūns |
melns |
|
PTD |
0,33 |
0,65 |
0,37 |
0,70 |
brūns |
oranžs |
violets* |
pelēks |
|
DAP |
0,07 |
– |
0 |
0,05 |
brūns* |
oranžs |
violets |
brūns |
|
H |
0,50 |
0,35 |
0,80 |
0,20 |
– |
oranžs |
violets |
melns* |
|
a·N |
0,90 |
0,80 |
0,90 |
0,75 |
oranži brūns |
– |
violets* |
melns |
|
0,37 |
– |
0,67 |
0,05 |
brūns |
ļoti bāli violets |
ļoti bāli brūns |
brūns* |
|
|
0,50 |
0,37 |
0,80 |
0,17 |
oranžs* |
bāli violets |
ļoti bāli brūns |
bāli brūns |
|
|
Piezīmes. 1. OPD parādās tikai nedaudz. Lai to skaidri atšķirtu no OTD, jāizmanto šķīdinātājs (atbilstoši šā pielikuma 122.22.3.apakšpunktam). 2. * Norāda labāko krāsu attīstījumu. |
||||||||
125. Pārbaudi divdimensiju plānslāņa hromatogrāfijā veic šādā kārtībā:
125.1. Divdimensiju hromatogrāfijas procedūrā izmanto šādus papildu standartus un reaģentus:
125.1.1. β·naftols (β·N).
125.1.2. 2-aminofenols (OAP).
125.1.3. 3-aminofenols (MAP).
125.1.4. 4-aminofenols (PAP).
125.1.5. 2-nitro-1,4-fenilēndiamīns (2-NPPD).
125.1.6. 4-nitro-1,2-fenilēndiamīns (4-NOPD).
No katra papildu standarta vai reaģenta sagatavo 0,5 % m/v šķīdumu saskaņā ar šī pielikuma 122.21.apakšpunktu.
125.2. Papildu attīstošais šķīdinātājs ir etilacetāta/cikloheksāna/amonjaka šķīdums, 25 % 65:30:0,5 (v/v/v).
125.3. Papildu indikatora sistēma: liek stikla trauku attīstīšanas kamerā plānslāņa hromatogrāfijai, pievieno apmēram 2 g kristāliskā joda un uzliek kamerai piemērotu vāku.
125.4. Hromatogrāfiju veic šādā kārtībā:
125.4.1. Uz plānslāņu plates (atbilstoši šā pielikuma 123.1.3.apakšpunktam) sorbenta virsmas uzzīmē divas līnijas (6.attēls).
125.4.2. Slāpekļa atmosfērā (atbilstoši šā pielikuma 123.1.1.apakšpunktam) 1–4 µl ekstrakta (atbilstoši šā pielikuma 124.1.apakšpunktam) klāj sākuma punktā 1 (6.attēls), kas ir 2 cm no abām malām. Ekstrakta daudzums atkarīgs no plankumu intensitātes šā pielikuma 124.2.apakšpunktā aprakstītajās hromatogrammās.
125.4.3. Starp punktu 2 un 3 (6.attēls) sadala oksidējošās krāsvielas, kas kvalitatīvi noteiktas vai ko uzskata par kvalitatīvi noteiktām (atbilstoši šā pielikuma 124.2.apakšpunktam) (attālums starp punktiem 1,5 cm). Klāj 2 µl katra standartšķīduma, izņemot DAP, – to klāj 6 µl. Šo darbību veic slāpeklī (atbilstoši šā pielikuma 125.4.2.apakšpunktam).
125.4.4. Sākuma punktā 4 un 5 (6.attēls) atkārto šā pielikuma 125.4.3.apakšpunktā minēto darbību un tur plati slāpeklī līdz hromatogrāfijas beigām (attālums starp punktiem 1,5 cm).
125.4.5. Izskalo hromatogrāfijas kameru ar slāpekli (atbilstoši šā pielikuma 122.8.apakšpunktam) un iepilda piemērotu daudzumu attīstošā šķīdinātāja (atbilstoši šā pielikuma 122.22.2.apakšpunktam). Liek plati (atbilstoši šā pielikuma 125.4.4.apakšpunktam) kamerā un tumsā attīsta pirmajā eluācijas virzienā (6.attēls). Eluē, līdz šķīdinātāja fronte ir sasniegusi uz plates atzīmēto līniju (apmēram 13 cm).
125.4.6. Izņem plati no kameras un liek hromatogrāfijas kamerā, kas iepriekš izskalota ar slāpekli, un vismaz 60 minūtes tvaicē eluentu.
125.4.7. Mēģenē ar iedaļām piemērotu daudzumu eluenta (atbilstoši šā pielikuma 125.2.apakšpunktam) ieliek kamerā, kas izskalota ar slāpekli (atbilstoši šā pielikuma 122.8.apakšpunktam). Ieliek kamerā (atbilstoši šā pielikuma 125.4.6.apakšpunktam) plati, kura pagriezta par 90°, un veic hromatogrāfiju otrā virzienā (arī tumsā), kamēr šķīdinātāja fronte sasniedz uz sorbenta virsmas uzzīmēto līniju. Izņem plati no kameras un iztvaicē eluentu gaisā.
125.4.8. Uz 10 minūtēm ieliek plati hromatogrāfijas kamerā ar joda tvaiku (atbilstoši šā pielikuma 125.3.apakšpunktam) un interpretē divdimensiju hromatogrammu, izmantojot vienlaicīgi hromatografēto standartvielu Rf vērtības un krāsas (Rf vērtības un krāsas norādītas šī pielikuma II tabulā). Lai iegūtu plankumu maksimālo krāsojumu, hromatogrammu pēc attīstīšanas pusstundu atstāj gaisā.
125.4.9. Saskaņā ar šā pielikuma 125.4.8.apakšpunktu noteikto oksidējošo krāsvielu klātbūtni pilnīgi var apstiprināt, atkārtojot šā pielikuma 125.4.1., 125.4.2., 125.4.3., 125.4.4., 125.4.5., 125.4.6., 125.4.7. un 125.4.8.apakšpunktā minētās darbības un sākuma punktā 1 pievienojot šā pielikuma 125.4.2.apakšpunktā minētajam ekstrakta daudzumam 1 µl šā pielikuma 125.4.8.apakšpunktā minēto standartvielu. Ja, salīdzinot ar hromatogrammu, kas iegūta saskaņā ar šā pielikuma 125.4.8.apakšpunktu, neatrod nevienu citu plankumu, šā pielikuma 125.4.8.apakšpunktā aprakstītā hromatogrammas interpretācija ir pareiza.
II tabula
Standartvielu krāsa pēc hromatogrāfijas un attīstīšanas ar joda tvaiku
|
Standartvielas |
Krāsa pēc attīstīšanas ar joda tvaiku |
|
R |
smilškrāsa |
|
P |
brūns |
|
a·N |
violets |
|
β·N |
bāli brūns |
|
H |
violeti brūns |
|
MPD |
dzeltenīgi brūns |
|
PPD |
violeti brūns |
|
MTD |
tumši brūns |
|
PTD |
dzeltenīgi brūns |
|
DAP |
tumši brūns |
|
OAP |
oranžs |
|
MAP |
dzeltenīgi brūns |
|
PAP |
violeti brūns |
|
2-NPPD |
brūns |
|
4-NOPD |
oranžs |
6.attēls
10. Nitrīta kvalitatīva un kvantitatīva noteikšana
10.1. Kvalitatīva noteikšana
126. Nitrīta kvalitatīvas noteikšanas metode nosaka procedūru nitrīta kvalitatīvai noteikšanai kosmētikas līdzekļos, īpaši krēmos un pastās.
127. Par nitrīta klātbūtni liecina krāsainu 2-aminobenzaldehīda fenilhidra-zonu (Nitrin ®) saturošu atvasinājumu veidošanās.
128. Nitrīta kvalitatīvai noteikšanai izmanto šādus reaģentus1:
128.1. Atšķaidīta sērskābe: atšķaida 2 ml koncentrētas sērskābes (d204 = 1,84) ar 11 ml destilēta ūdens.
128.2. Atšķaidīta sālsskābe: atšķaida 1 ml koncentrētas sālsskābes (d204 = 1,19) ar 11 ml destilēta ūdens.
128.3. Metanols.
128.4. 2-aminobenzaldehīda fenilhidrazona (Nitrin ® reaģenta) šķīdums metanolā:
128.4.1. Nosver 2,0 g Nitrin ® un kvantitatīvi pārnes uz 100 ml mērkolbu.
128.4.2. Pa pilienam pievieno 4 ml atšķaidītas sālsskābes (atbilstoši šā pielikuma 128.2.apakšpunktam) un sakrata.
128.4.3. Uzpilda līdz zīmei ar metanolu un maisa, līdz šķīdums ir pilnīgi dzidrs.
128.4.4. Šķīdumu glabā tumšā stikla pudelē (atbilstoši šā pielikuma 129.3.apakšpunktam).
129. Nitrīta kvalitatīvai noteikšanai izmanto šādu iekārtu:
129.1. Vārglāzes, 50 ml.
129.2. Mērkolba, 100 ml.
129.3. Tumša stikla pudele, 125 ml.
129.4. Stikla plate, 10 × 10 cm.
129.5. Plastikas karotīte.
129.6. Filtrpapīrs, 10 × 10 cm.
130. Nitrīta kvalitatīvu noteikšanu veic šādā kārtībā:
130.1. Daļu analizējamā parauga vienmērīgi izlīdzina uz stikla plates (atbilstoši šā pielikuma 129.4.apakšpunktam) kārtā, kas nav biezāka par 1 cm.
130.2. Samērcē filtrpapīra (atbilstoši šā pielikuma 129.6.apakšpunktam) loksni destilētā ūdenī. To uzliek uz parauga un piespiež ar plastikas karotīti.
130.3. Pagaida apmēram vienu minūti un filtrpapīra centrā, uzpilina divus pilienus atšķaidītas sērskābes (atbilstoši šā pielikuma 128.1.apakšpunktam), pēc tam divus pilienus Nitrin ® šķīduma (atbilstoši šā pielikuma 128.4.apakšpunktam).
130.4. Pēc 5–10 sekundēm noņem filtrpapīru un apskata pret dienasgaismu. Par nitrīta klātbūtni liecina purpursarkans krāsojums. Ja nitrīta saturs ir zems, purpursarkanais krāsojums 5–15 sekundēs kļūst dzeltens. Ja nitrīta daudzums ir liels, krāsa mainās tikai pēc vienas līdz divām minūtēm. Purpursarkanās krāsas intensitāte un laiks, kādā tā kļūst dzeltena, var liecināt par nitrīta saturu paraugā.
10.2. Kvantitatīva noteikšana
131. Ar nitrīta kvantitatīvās noteikšanas metodi kvantitatīvi nosaka nitrītu kosmētikas līdzekļos.
132. Ar nitrīta kvantitatīvās noteikšanas metodi noteikto nitrīta saturu izsaka nātrija nitrīta masas procentos.
133. Kad paraugs ir atšķaidīts ar ūdeni un dzidrināts, nitrītam liek reaģēt ar sulfanilamīdu un N-1-naftiletilēndiamīnu un 538 nm līmenī izmēra iegūtās krāsas blīvumu.
134. Nitrīta kvantitatīvās noteikšanas metodei izmanto šādus reaģentus1:
134.1. Dzidrināšanas reaģenti (nedrīkst izmantot ilgāk kā vienu nedēļu pēc sagatavošanas):
134.1.1. I Karesa reaģents: izšķīdina 106 g kālija cianoferāta(II) K4Fe(CN)6 × 3H2O destilētā ūdenī un atšķaida ar ūdeni līdz 1000 ml.
134.1.2. II Karesa reaģents: izšķīdina 219,5 g cinka acetāta Zn(CH3COO)2 × 2H2O un 30 ml ledus etiķskābes destilētā ūdenī un atšķaida ar ūdeni līdz 1000 ml.
134.2. Nātrija nitrīta šķīdums: izšķīdina 0,500 g nātrija nitrīta destilētā ūdenī 1000 ml mērkolbā un atšķaida ar ūdeni līdz zīmei. Atšķaida 10,0 ml šī standartšķīduma līdz 500 ml. 1 ml pēdējā šķīduma ir vienāds ar 10 mikrogramiem NaNO2.
134.3. 1 n nātrija hidroksīda šķīdums.
134.4. 0,2 % sulfanilamīda hidrohlorīda šķīdums: sildot izšķīdina 2,0 g sulfanilamīda 800 ml ūdens. Atdzesē un maisot pievieno 100 ml koncentrētas sālsskābes. Atšķaida ar ūdeni līdz 1000 ml.
134.5. 5 n sālsskābe.
134.6. N-1-naftilreaģents: gatavo izmantošanas dienā. Izšķīdina 0,1 g N-1-naftiletilēndiamīna dihidrohlorīda ūdenī un atšķaida ar ūdeni līdz 100 ml.
135. Nitrīta kvantitatīvās noteikšanas metodei izmanto šādu iekārtu:
135.1. Analītiskie svari.
135.2. Mērkolbas, 100, 250, 500 un 1000 ml.
135.3. Mērpipetes vai pipetes ar iedaļām.
135.4. Mērcilindri, 100 ml.
135.5. Kroku filtrpapīri bez nitrītiem, ar diametru 15 cm.
135.6. Ūdens vanna.
135.7. Spektrofotometrs ar 1 cm garām kivetēm.
135.8. pH metrs.
135.9. Mikrobirete, 10 ml.
135.10. Vārglāzes, 250 ml.
136. Nitrīta kvantitatīvo noteikšanu veic šādā kārtībā:
136.1. Ar 0,1 mg precizitāti nosver aptuveni 0,5 g (turpmāk - m grami) homogenizētā parauga, ar karstu destilētu ūdeni kvantitatīvi pārnes uz 250 ml vārglāzi un uzpilda apmēram līdz 150 ml ar karstu destilētu ūdeni. Uz pusstundu liek vārglāzi ūdens vannā 80 °C temperatūrā. Šajā laikā ik pa brīdim sakrata.
136.2. Atdzesē līdz istabas temperatūrai un maisot secīgi pievieno 2 ml I Karesa reaģenta (atbilstoši šā pielikuma 134.1.1.apakšpunktam) un 2 ml II Karesa reaģenta (atbilstoši šā pielikuma 134.1.2.apakšpunktam).
136.3. Pievieno 1 n nātrija hidroksīda šķīdumu, lai noregulētu pH uz 8,3 (izmanto pH metru). Saturu kvantitatīvi pārnes uz 250 ml mērkolbu un uzpilda līdz zīmei ar destilētu ūdeni.
136.4. Samaisa un izfiltrē caur kroku filtrpapīru (atbilstoši šā pielikuma 135.5.apakšpunktam).
136.5. Ar pipeti (atbilstoši šā pielikuma 135.3.apakšpunktam) piemērotu alikvotu (turpmāk -V ml) daļu, bet ne vairāk kā 25 ml, dzidrā filtrāta iepilina 100 ml mērkolbā un pievieno destilētu ūdeni līdz 60 ml tilpumam.
136.6. Samaisa, pievieno 10,0 ml sulfanilamīda hidrohlorīda šķīduma (atbilstoši šā pielikuma 134.4.apakšpunktam) un pēc tam 6,0 ml 5 n sālsskābi (atbilstoši šā pielikuma 134.5.apakšpunktam). Sajauc un piecas minūtes ļauj nostāties. Pievieno 2,0 ml N-1-naftilreaģenta (atbilstoši šā pielikuma 134.6.apakšpunktam), samaisa un trīs minūtes ļauj nostāties. Atšķaida ar ūdeni līdz atzīmei un samaisa.
136.7. Sagatavo tukšo analīzi, atkārtojot šā pielikuma 136.5. un 136.6.apakšpunktā minētās darbības bez N-1-naftilreaģenta.
136.8. Izmēra (atbilstoši šā pielikuma 135.7.apakšpunktam) saskaņā ar šā pielikuma 136.6.apakšpunktu iegūtā šķīduma optisko blīvumu 538 nm līmenī, par standartu izmantojot tukšo šķīdumu (atbilstoši šā pielikuma 136.7.apakšpunktam).
136.9. No kalibrēšanas līknes (atbilstoši šā pielikuma 136.10.apakšpunktam) nolasa nātrija nitrīta saturu mikrogramos uz 100 ml šķīduma (turpmāk - m1mikrogrami), kas atbilst optiskajam blīvumam, kuru mēra saskaņā ar šā pielikuma 136.8.apakšpunktu.
136.10. Izmantojot 10 μg uz ml nātrija nitrīta šķīduma (atbilstoši šā pielikuma 134.2.apakšpunktam), zīmē kalibrēšanas līkni nātrija nitrītam 0, 20, 40, 60, 80, 100 µg koncentrācijā.
137. Aprēķina nātrija nitrīta saturu paraugā masas procentos pēc formulas:
% NaNO2 = ((250 / V) × m1 × 10-6 × (100 / m)) = ((m1) / (V × m × 40)), kur
m – noteikšanai ņemtā parauga (atbilstoši136.1.apakšpunktam) masa (gramos);
m1 – nātrija nitrīta saturs (mikrogramos), kas noteikts saskaņā ar šā pielikuma 136.9.apakšpunktu;
V – mērījumam izmantotais filtrāts (mililitros) (atbilstoši šā pielikuma 136.5.apakšpunktam).
138. No viena parauga ar aptuveni 0,2 % (m/m) nātrija nitrīta saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,005 % vērtību4.
11. Brīvā formaldehīda kvalitatīva un kvantitatīva noteikšana
139. Brīvā formaldehīda kvalitatīvas un kvantitatīvas noteikšanas metodi izmanto, lai noteiktu kvalitatīvo un kvantitatīvo formaldehīda donoru klātbūtni vai neesību. Tā ir piemērojama visiem kosmētikas līdzekļiem.
140. Kvantitatīvu noteikšanu ar pentān-2,4-diona kolorimetriju piemēro, ja formaldehīdu izmanto vienu pašu vai kopā ar citiem konservantiem, kas nav formaldehīda donori. Šajā metodē atvasināšanas laikā formaldehīda donori atšķeļas un dod pārāk augstus rezultātus (saistītais un polimērais formaldehīds). Šķidrumu hromatogrāfijā jāatdala brīvais formaldehīds.
141. Kvantitatīvi noteikta brīva formaldehīda saturu paraugā izsaka masas procentos.
11.1. Kvalitatīva noteikšana
142. Brīvais un saistītais formaldehīds sērskābes vidē Šifa reaģentu nokrāso rozā vai sarkanīgi violetu.
143. Brīvā formaldehīda kvalitatīvas noteikšanas metodei izmanto šādus reaģentus1:
143.1. Fuksīns.
143.2. Nātrija sulfīts, hidratēts līdz 7H2O.
143.3. Koncentrēta sālsskābe (d=1,19).
143.4. Sērskābe, aptuveni 1 M.
143.5. Šifa reaģents: 100 mg fuksīna iesver vārglāzē un 80 °C izšķīdina 75 ml ūdens. Pēc atdzesēšanas pievieno 2,5 g nātrija sulfīta heptahidrāta (Na2SO3·7H2O) un 1,5 ml koncentrētas sālsskābes (d204 = 1,19). Uzpilda līdz 100 ml (pēc divām nedēļām šis reaģents vairs nav izmantojams).
144. Brīvā formaldehīda kvalitatīvu noteikšanu veic šādā kārtībā:
144.1. Iesver 2 g analizējamā parauga 10 ml vārglāzē.
144.2. Piepilina divus pilienus 2 N sērskābes un 2 ml Šifa reaģenta (šim reaģentam izmantošanas laikā jābūt pilnīgi bezkrāsainam). Sakrata un atstāj uz piecām minūtēm.
144.3. Ja šā pielikuma 144.2.apakšpunktā minētajās piecās minūtēs novēro rozā vai sarkanīgi violetu krāsojumu, šķīdumā ir formaldehīds koncentrācijā, kas pārsniedz 0,01 %, un to nosaka kvantitatīvi, veicot šā pielikuma 11.2.apakšnodaļā minēto brīvo un saistīto metodi un, ja nepieciešams, šā pielikuma 11.3.apakšnodaļā minēto procedūru.
11.2. Vispārēja kvantitatīva noteikšana ar pentān-2,4-diona kolorimetriju
145. Formaldehīds amonija acetāta klātbūtnē reaģē ar pentān-2,4-dionu, veidojot 3,5-diacetil-1,4-dihidrolutidīnu. To ekstrahē ar butān-1-olu un izmēra ekstrakta absorbciju 410 nm līmenī.
146. Vispārējai kvantitatīvai noteikšanai ar pentān-2,4-diona kolorimetriju izmanto šādus reaģentus1:
146.1. Bezūdens amonija acetāts.
146.2. Koncentrēta etiķskābe, d204 = 1,05.
146.3. Pentān-2,4-dions (tikko destilēts pazeminātā spiedienā 25 mm Hg 25° – tam nebūtu jāabsorbē pie 410 nm).
146.4. Butān-1-ols.
146.5. Sālsskābe, 1 M.
146.6. Sālsskābe, aptuveni 0,1 M.
146.7. Nātrija hidroksīds, 1 M.
146.8. Izšķīdināta ciete, tikko pagatavota saskaņā ar Eiropas farmakopeju (1 g/50 ml ūdens), 2.izdevums, 1980.gads, I-VII-1-1 daļa.
146.9. 37–40 % (m/v) formaldehīda.
146.10. Joda standartšķīdums, 0,05 M.
146.11. Nātrija tiosulfāta standartšķīdums, 0,1 M.
146.12. Pentān-2,4-diona reaģents (reaģentu sagatavo īsi pirms lietošanas):
146.12.1. Mērkolbā ar 1000 ml ietilpību izšķīdina 150 g amonija acetāta, 2 ml pentān-2,4-diona (tikko sagatavota, destilēta pazeminātā spiedienā – tam nebūtu jāabsorbē pie 410 nm) un 3 ml ledus etiķskābes.
146.12.2. Uzpilda ar ūdeni līdz 1000 ml (šķīduma pH aptuveni 6,4).
146.13. Reaģents (atbilstoši šā pielikuma 146.12.apakšpunktam) bez pentān-2,4-diona.
146.14. Formaldehīda references standarts: standartšķīdums:
146.14.1. Ielej 5 g 37–40 % formaldehīda šķīduma 1000 ml mērkolbā un uzpilda līdz 1000 ml.
146.14.2. Šī šķīduma stiprumu nosaka šādi: nolej 10,00 ml, pievieno 25,00 ml 0,1 n joda standartšķīduma un 10 ml nātrija hidroksīda šķīduma.
146.14.3. Ļauj nostāties piecas minūtes.
146.14.4. Pievieno 11 ml HCl un kvantitatīvi nosaka joda pārākumu ar 0,1 n nātrija tiosulfāta standartšķīdumu, par indikatoru izmantojot cietes šķīdumu. 1 ml 0,05 M joda šķīduma, ko patērē, ir līdzvērtīgs 1,5 mg formaldehīda.
146.15. Formaldehīda references standarts: atšķaidīts šķīdums: formaldehīda standartšķīdumu secīgi atšķaida ar ūdeni 1/20 un tad 1/100. 1 ml šī šķīduma satur apmēram 1mg formaldehīda. Aprēķina reālo daudzumu.
147. Vispārējai kvantitatīvai noteikšanai ar pentān-2,4-diona kolorimetriju izmanto šādu iekārtu:
147.1. Standarta laboratorijas iekārta.
147.2. Fāzu atdalīšanas filtrs, vatmaņpapīrs 1PS (vai līdzvērtīgs).
147.3. Centrifūga.
147.4. Ūdens vanna, kurā temperatūra noregulēta uz 60 °C.
147.5. Spektrofotometrs.
147.6. Stikla kivetes ar optiskā ceļa garumu 1 cm.
148. Vispārēju kvantitatīvu noteikšanu ar pentān-2,4-diona kolorimetriju veic šādā kārtībā:
148.1. Parauga šķīdums. 100 ml mērkolbā ar 0,001 g precizitāti iesver analizējamā parauga daudzumu gramos, kas atbilst iepriekš pieņemtam aptuvenam 150 mikrogramu formaldehīda daudzumam. Uzpilda ar demineralizētu ūdeni līdz 100 ml un samaisa (turpmāk -S šķīdums) (pārbauda, lai pH būtu tuvu 6 pH. Ja tas tā nav, atšķaida sālsskābes šķīdumā). Ērlenmeijera kolbā ar 50 ml ietilpību pievieno 10,00 ml S šķīduma, 5,00 ml pentān-2,4-diona reaģenta un deminerlizētu ūdeni līdz galīgajam 30 ml tilpumam.
148.2. Kontrolšķīdums. Ar kontrolšķīdumu analizējamā paraugā novērš iespējamu traucējošu fona krāsas iedarbību. Ērlenmeijera 50 ml kolbā pievieno 10,00 ml S šķīduma, 5,00 ml reaģenta (atbilstoši šā pielikuma 146.13.apakšpunktam) un demineralizētu ūdeni līdz galīgajam 30 ml tilpumam.
148.3. Tukšā analīze. Ērlenmeijera 50 ml kolbā pievieno 5,00 ml pentān-2,4-diona reaģenta (atbilstoši šā pielikuma 146.12.apakšpunktam), papildina ar demineralizētu ūdeni līdz 30 ml.
148.4. Kolbas (atbilstoši šā pielikuma 148.1., 148.2. un 148.3.apakšpunktam) sakrata un tieši uz 10 minūtēm iegremdē ūdens vannā 60 °C. Ļauj atdzist divas minūtes vannā ar ledus ūdeni.
148.5. Pārnes uz 50 ml dalāmajām piltuvēm, kurās ir 10,00 ml butān-1-ola. Katru kolbu izskalo ar 3–5 ml ūdens un saskalojumus pievieno piltuvju saturam. Tieši 30 sekundes maisījumu stipri krata. Ļauj tam sadalīties.
148.6. Iefiltrē kivetēs caur fāžu atdalīšanas filtru. Var centrifugēt (3000 apgr./min 5 minūtes).
148.7. Izmēra šā pielikuma 148.1.apakšpunktā norādītā parauga šķīduma ekstrakta absorbciju A1 410 nm līmenī un salīdzina ar kontrolšķīduma (atbilstoši šā pielikuma 148.2.apakšpunktam) ekstraktu.
148.8. Izmēra tukšā šķīduma ekstrakta absorbciju A2 un salīdzina ar butān-1-olu (visas šā pielikuma 148.4, 148.5., 148.6. un 148.7.apakšpunktā minētās darbības jāveic 25 minūtēs, skaitot no brīža, kad Ērlenmeijera kolbas ieliek ūdens vannā 60 °C) .
149. Kalibrēšanas līkni konstruē šādi:
149.1. Ērlenmeijera 50 ml kolbā ielej 5,00 ml standartšķīduma (atbilstoši šā pielikuma 146.15.apakšpunktam), 5,00 ml pentān-2,4-diona reaģenta (atbilstoši šā pielikuma 146.12.apakšpunktam), uzpilda līdz 30 ml ar demineralizētu ūdeni.
149.2. Turpina saskaņā ar šā pielikuma 148.4., 148.5., 148.6., 148.7 un 148.8.apakšpunktu, izmēra absorbciju, salīdzinot to ar butān-1-olu.
149.3. Atkārto procedūru ar 10, 15, 20 un 25 ml atšķaidīta standartšķīduma (atbilstoši šā pielikuma 146.15.apakšpunktam).
149.4. Nullpunkta vērtību iegūst saskaņā ar šā pielikuma 148.8.apakšpunktā minēto kārtību.
149.5. Kalibrēšanas līkni konstruē, atņemot nullpunkta vērtību no katras absorbcijas, kas iegūta saskaņā ar šā pielikuma 149.1. un 149.3.apakšpunktu. Bēra likums ir spēkā, ja formaldehīds nepārsniedz 30 µg.
150. Aprēķinus veic šādā kārtībā:
150.1. Atņem A2 no A1 un nolasa no kalibrēšanas līknes formaldehīda daudzumu C mikrogramos analizējamā šķīdumā.
150.2. Parauga formaldehīda saturu masas procentos (% m/m) aprēķina pēc šādas formulas:
formaldehīda saturs % = ((C) / (103 × m)), kur
m – testa porcijas masa (gramos).
151. Ja formaldehīda saturs kosmētikas līdzeklī ir 0,2 %, no viena parauga divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,005 %, nosakot ar pentān-2,4-diona kolorometriju.
152. Saskaņā ar šī pielikuma 11.3.apakšnodaļā minēto darba gaitu rīkojas tad, ja brīvā formaldehīda kvantitatīvajā noteikšanā iegūst rezultātus, kas ir lielāki par šajos noteikumos noteiktajām maksimālajām koncentrācijām:
152.1. 0,05–0,2 % nemarķētā produktā.
152.2. Produktā, kas ir vai nav marķēts, koncentrācija ir lielāka par 0,2 %.
11.3. Kvantitatīva noteikšana formaldehīda donoru klātbūtnē
153. Atdalīto formaldehīdu pārnes dzeltenā lutidīna atvasinājumā, izmantojot reakciju ar pentān-2,4-dionu pēckolonnas reaktorā, un iegūto atvasinājumu nosaka pēc absorbcijas 420 nm līmenī.
154. Kvantitatīvai noteikšanai formaldehīda donoru klātbūtnē izmanto šādus reaģentus1, 3:
154.1. Bezūdens amonija acetāts.
154.2. Koncentrēta etiķskābe.
154.3. Pentān-2,4-dions (glabāts 4 °C).
154.4. Bezūdens dinātrija fosfāts.
154.5. 85 % ortofosforskābe (d = 1,7).
154.6. Metanols2.
154.7. Dihlormetāns.
154.8. 37–40 % V formaldehīds.
154.9. Nātrija hidroksīds, 1 M.
154.10. Sālsskābe, 1 M.
154.11. Sālsskābe, 0,002 M.
154.12. Tikko pagatavota izšķīdināta ciete (atbilstoši šā pielikuma 146.8.apakšpunktam).
154.13. Joda standartšķīdums, 0,05 M.
154.14. Nātrija tiosulfāta standartšķīdums, 0,1 M.
154.15. Kustīgā fāze: nātrija fosfāta ūdens šķīdums (atbilstoši šā pielikuma 154.4.apakšpunktam), 0,006 M, kas koriģēts līdz pH 2,1 ar ortofosforskābi (atbilstoši šā pielikuma 154.5.apakšpunktam).
154.16. Pēckolonnas reaģents: mērkolbā ar 1000 ml ietilpību izšķīdina 62,5 g amonija acetāta (atbilstoši šā pielikuma 154.1.apakšpunktam), 7,5 ml etiķskābes (atbilstoši šā pielikuma 154.2.apakšpunktam) un 5 ml pentān-2,4-diona (atbilstoši šā pielikuma 154.3.apakšpunktam). Uzpilda ar ūdeni līdz 1000 ml. Reaģentu tur tumšā vietā. Maksimālais uzglabāšanas laiks ir trīs dienas 25 ºC temperatūrā. Nedrīkstētu būt krāsas izmaiņu.
154.17. Formaldehīda references standarts: standartšķīdums:
154.17.1. Ielej 10 g 37–40 % formaldehīda šķīduma 1000 ml mērkolbā un uzpilda līdz 1000 ml.
154.17.2. Formaldehīda šķīduma stiprumu nosaka šādi: nolej 5,00 ml; pievieno 25,00 ml joda standartšķīduma un 10 ml nātrija hidroksīda šķīduma.
154.17.3. Ļauj nostāties piecas minūtes.
154.17.4. Pievieno 11 ml HCl un kvantitatīvi nosaka joda pārākumu ar 0,1 n nātrija tiosulfāta standartšķīdumu, par indikatoru izmantojot cietes šķīdumu. 1 ml 0,05 M joda šķīduma, ko patērē, ir līdzvērtīgs 1,5 mg formaldehīda.
154.18. Formaldehīda references standarts: atšķaidīts šķīdums. Formaldehīda standartšķīdumu atšķaida līdz 1/100 tā izejas šķīduma kustīgajā fāzē (atbilstoši šā pielikuma 154.15.apakšpunktam). 1 ml šī šķīduma satur apmēram 37 mg formaldehīda. Aprēķina reālo daudzumu.
155. Kvantitatīvai noteikšanai formaldehīda donoru klātbūtnē izmanto šādu iekārtu:
155.1. Standarta laboratorijas iekārta.
155.2. HPCL sūknis, bez vibrācijas.
155.3. Zemspiediena, nevibrējošs sūknis reaģentam (vai otrs HPLC sūknis).
155.4. Inžektora vārsts ar 10 ml cilpu.
155.5. Pēckolonnas reaktors ar šādām sastāvdaļām:
155.5.1. Viena 1 litra trīskaklu kolba.
155.5.2. Viens 1 litra kolbas sildītājs.
155.5.3. Divas Vigrē kolonnas ar vismaz 10 šķīvjiem, no kuriem divi ir dzesējami ar gaisu.
155.5.4. Nerūsējošā tērauda caurule (siltuma apmaiņai) 1,6 mm, iekšējais diametrs 0,23 mm, garums 400 mm.
155.5.5. Teflona caurule 1,6 mm, iekšējais diametrs 0,30 mm, garums 5 m (reakcijas cilpa).
155.5.6. Viena T-veida daļa bez mirušā tilpuma (Valko vai līdzvērtīga).
155.5.7. Trīs savienojumi bez mirušā tilpuma vai viena pēckolonnas uzmava Applied Biosystems PCRS 520 vai līdzvērtīga, kas aprīkota ar 1 ml reaktoru.
155.6. Membrānfiltrs, kura poru lielums ir 0,45 mm.
155.7. SEP-PAKR C18 kārtridžs vai līdzvērtīgs.
155.8. Gatavas kolonnas: Bischoff hypersil RP 18 (veida NC norāde C 25.46 1805) (5 mm, garums 250 mm, iekšējais diametrs 4,6 mm) vai Dupont, Zorbax ODS (5 mm, garums 250 mm, iekšējais diametrs 4,6 mm), vai Phase SEP, spherisorb ODS 2 (5 mm, garums 250 mm, iekšējais diametrs 4,6 mm).
155.9. Pirmskolonna Bischoff K1 hypersil RP 18 (norāde K1 G 6301 1805) (5 mm, garums 10 mm, vai līdzvērtīga).
155.10. Kolonna un priekškolonna ir savienotas ar Ecotube sistēmu (norāde A 15020508 Bischoff) vai līdzvērtīgu.
155.11. Ierīci (atbilstoši šā pielikuma 155.5.apakšpunktam) komplektē, savienojumus pēc iešprices tilpuma saglabājot iespējami īsu brīdi. Šādā gadījumā nerūsējošā tērauda caurule starp reaktora izeju un detektora ieeju ir paredzēta maisījuma atdzesēšanai pirms kvantitatīvas noteikšanas, un temperatūra detektorā nav zināma, bet ir nemainīga.
155.12. Redzams UV detektors.
155.13. Pašrakstītājs.
155.14. Centrifūga.
155.15. Ultraskaņas vanna.
155.16. Vibrācijas tipa maisītājs (virpuļmaisītājs vai līdzvērtīgs).
156. Kvantitatīvu noteikšanu formaldehīda donoru klātbūtnē veic šādā kārtībā:
156.1. Kalibrēšanas līkne. To iegūst, uzzīmējot pīķa augstumus, kas ir formaldehīda references standarta – atšķaidīta – koncentrācijas funkcija. Sagatavo standartšķīdumus, formaldehīda standartšķīdumu (atbilstoši šā pielikuma 154.18.apakšpunktam) atšķaidot ar kustīgo fāzi (atbilstoši šā pielikuma 154.15.apakšpunktam):
156.1.1. 1,00 ml šķīduma (atbilstoši šā pielikuma 154.18.apakšpunktam), ko atšķaida līdz 20,00 ml (aptuveni 185 mg/100 ml).
156.1.2. 2,00 ml šķīduma (atbilstoši šā pielikuma 154.18.apakšpunktam), ko atšķaida līdz 20,00 ml (aptuveni 370 mg/100 ml).
156.1.3. 5,00 ml šķīduma (atbilstoši šā pielikuma 154.18.apakšpunktam), ko atšķaida līdz 25,00 ml (aptuveni 740 mg/100 ml).
156.1.4. 5,00 ml šķīduma (atbilstoši šā pielikuma 154.18.apakšpunktam), ko atšķaida līdz 20,00 ml (aptuveni 925 mg/100 ml).
Šī pielikuma 156.1.1., 156.1.2., 156.1.3. un 156.1.4.apakšpunktā minētos standartšķīdumus nostādina vienu stundu laboratorijas temperatūrā, un tiem jābūt tikko pagatavotiem. Kalibrācijas līknes linearitāte ir laba koncentrācijām no 1,00 līdz 15,00 mg/ml.
156.2. Paraugu pagatavošanu veic šādā kārtībā:
156.2.1. Emulsijas (krēmi, grims, kontūrzīmuļi). Aizkorķētā 100 ml kolbā ar 0,001 g precizitāti iesver analizējamā parauga daudzumu (mg), kas atbilst iepriekš pieņemtam 100 mg formaldehīda daudzumam. Pievieno precīzi nomērītus 20,00 ml dihlormetāna un 20,00 ml sālsskābes. Sajauc ar vibrācijas maisītāju, izmantojot ultraskaņas vannu. Centrifugējot sadala pa divām fāzēm (3000 g divas minūtes). Tikmēr ar 2 ml metanola nomazgā kārtridžu, pēc tam kondicionē ar 5 ml ūdens. 4 ml ekstrakta ūdens fāzes iztecina cauri kondicionētajam kārtridžam, pirmos 2 ml izlej un nākamo daļu atgūst.
156.2.2. Losjoni, šampūni. Aiztaisītā 100 ml kolbā ar 0,001 g precizitāti iesver analizējamā parauga daudzumu (mg), kas atbilst iepriekš pieņemtam aptuvenam 500 mg formaldehīda daudzumam. Uzpilda līdz 100 ml ar kustīgo fāzi (atbilstoši šā pielikuma 154.15.apakšpunktam). Šo šķīdumu izfiltrē caur filtru (atbilstoši šā pielikuma 155.6.apakšpunktam) un iešļircina vai izlaiž caur kārtridžu (atbilstoši šā pielikuma 155.7.apakšpunktam), kas kondicionēts šī pielikuma 156.2.1.apakšpunktā minētajā veidā. Visus šķīdumus iešļircina tūlīt pēc pagatavošanas.
156.3. Hromatogrāfijas nosacījumi:
156.3.1. Kustīgās fāzes plūsmas ātrums 1 ml/min.
156.3.2. Reaģenta plūsmas ātrums 0,5 ml/min.
156.3.3. Kopējais plūsmas ātrums pie detektora izejas 1,5 ml/min.
156.3.4. Iešprices daudzums 10 ml.
156.3.5. Eluācijas temperatūra. Ja viela grūti sadalās pa fāzēm, iegremdē kolonnu vannā ar kūstošu ledu un gaida, kamēr temperatūra nostabilizējas (15–20 min).
156.3.6. Pēckolonnas reakcijas temperatūra 100 °C.
156.3.7. Kvantitatīva noteikšana 420 nm līmenī.
157. Visa hromatogrāfijas sistēma un pēckolonna pēc lietošanas jāskalo ar ūdeni. Ja sistēmu nelieto ilgāk par divām dienām, pēc skalošanas ar ūdeni jāskalo ar metanolu. Pirms sistēmas rekondicionēšanas caur to iztecina ūdeni, lai izvairītos no rekristalizācijas.
158. Aprēķina šādi:
Emulsijas – saskaņā ar šī pielikuma 156.2.1.apakšpunktu.
Formaldehīda % saturs (m/m):
Losjoni, šampūni
Šajā fāzē ir šāda formula:
![]() , kur
, kur
m – analizētā parauga masa (gramos) (atbilstoši šā pielikuma 156.2.1.apakšpunktam);
C – formaldehīda koncentrācija mg/100 ml, ko nolasa no kalibrēšanas līknes (atbilstoši šā pielikuma 156.1.apakšpunktam).
159. No viena parauga ar 0,05 % formaldehīda saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpība nedrīkstētu pārsniegt 0,001 %. No viena parauga ar 0,2 % formaldehīda saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpība nedrīkstētu pārsniegt 0,005 %, kas ir vienāds ar 1,5 mg formaldehīda4.
12. Rezorcīna kvantitatīva noteikšana šampūnos un matu losjonos
160. Ar rezorcīna kvantitatīvas noteikšanas metodi gāzu hromatogrāfijā kvantitatīvi nosaka rezorcīnu šampūnos un matu losjonos. Metode izmantojama paraugiem ar koncentrāciju no 0,1 līdz 2,0 masas %.
161. Kvantitatīvi noteiktā rezorcīna saturu paraugā izsaka masas procentos.
162. Rezorcīnu un 3,5-dihidroksitoluolu, (5-metilrezorcīns), kas pievienots kā iekšējais standarts, atdala no parauga plānslāņa hromatogrāfijā. Abus savienojumus izolē, noskrāpējot to plankumus no plānslāņu plates un ekstrahējot ar metanolu. Beidzot ekstrahētos savienojumus izžāvē, sililē un kvantitatīvi nosaka gāzu hromatogrāfijā.
163. Rezorcīna kvantitatīvai noteikšanai izmanto šādus reaģentus1:
163.1. Sālsskābe, 25 % (m/m).
163.2. Metanols.
163.3. Etanols, 96 % (v/v).
163.4. Izmantošanai gatavas silikagela TLC loksnes (plastikas vai alumīnija) ar fluorescentu indikatoru. Deaktivē šādi:
163.4.1. Apsmidzina parastās iepriekš pārklātās silīcija oksīda loksnes ar ūdeni, līdz izveidojas glazūrveida pārklājums.
163.4.2. Apsmidzinātajām platēm ļauj žūt gaisā istabas temperatūrā vienu līdz trīs stundas. Ja plates nav deaktivētas, neatgriezeniska adsorbcija silīcija oksīdā var radīt rezorcīna zudumus.
163.5. Attīstošais šķīdinātājs: acetons/hloroforms/etiķskābe 20:75:5 (v/v/v).
163.6. Rezorcīna standartšķīdums: izšķīdina 400 mg rezorcīna 100 ml 96 % etanola (atbilstoši šā pielikuma 163.3.apakšpunktam) (1 ml atbilst 4000 µg rezorcīna).
163.7. Iekšējais standartšķīdums: izšķīdina 400 mg 3,5-dihidroksitoluola (turpmāk - DHT) 100 ml 96 % etanola (1 ml atbilst 4000 µg DHT).
163.8. Standartmaisījums: 100 ml mērkolbā samaisa 10 ml šķīduma, kas norādīts šā pielikuma 163.6.apakšpunktā, un 10 ml šķīduma, kas norādīts šā pielikuma 163.7.apakšpunktā, uzpilda līdz zīmei ar 96 % etanolu un samaisa (1 ml atbilst 400 µg rezorcīna un 400 µg DHT).
163.9. Sililējošie aģenti:
163.9.1. N,O-bis-(trimetilsilil)trifluoracetamīds (BSTFA).
163.9.2. Heksametildisilazāns (HMDS).
163.9.3. Trimetilhlorsilāns (TMCS).
164. Rezorcīna kvantitatīvai noteikšanai izmanto šādu iekārtu:
164.1. Standarta plānslāņu un gāzu hromatogrāfijas iekārta.
164.2. Stikla trauki.
165. Rezorcīna kvantitatīvu noteikšanu veic šādā kārtībā:
165.1. Parauga sagatavošana:
165.1.1. Precīzi iesver 150 ml vārglāzē analizējamo kosmētikas līdzekļa daudzumu (m grami), kas satur aptuveni 20–50 mg rezorcīna.
165.1.2. Skābina ar sālsskābi (atbilstoši šā pielikuma 163.1.apakšpunktam), līdz maisījums ir skābs (vajadzīgi aptuveni 2–4 ml), pievieno 10 ml (40 mg DHT) iekšējā standartšķīduma (atbilstoši šā pielikuma 163.7.apakšpunktam) un samaisa. Pārnes uz 100 ml mērkolbu ar etanolu (atbilstoši šā pielikuma 163.3.apakšpunktam), uzpilda līdz zīmei ar etanolu un samaisa.
165.1.3. Apmēram 8 cm garā nepārtrauktā līnijā klāj 250 ml šķīduma (atbilstoši šā pielikuma 165.1.2.apakšpunktam) uz deaktivētas silīcija oksīda loksnes (atbilstoši šā pielikuma 163.4.apakšpunktam). Līniju veido iespējami šauru.
165.1.4. Saskaņā ar šā pielikuma 165.1.3.apakšpunktu uz tās pašas plates klāj 250 ml standartmaisījuma (atbilstoši šā pielikuma 163.8.apakšpunktam).
165.1.5. Divos punktos uz starta līnijas liek 5 ml no katra šķīduma, kas norādīts šā pielikuma 163.6. un 163.7.apakšpunktā, lai pēc attīstīšanas atvieglotu lokalizāciju.
165.1.6. Attīsta plati nepiesātinātā kamerā, kas piepildīta ar attīstošo šķīdinātāju, kurš norādīts šā pielikuma 163.5.apakšpunktā, līdz šķīdinātāja fronte ir sasniegusi līniju, kas atrodas 12 cm no starta līnijas. Parasti tam vajadzīgas 45 minūtes. Nožāvē plati un UV gaismā (254 nm) nosaka rezorcīna/DHT zonu. Abiem savienojumiem ir aptuveni vienāda Rf vērtība. Ar zīmuli apvelk 2 mm attālumā no ārējās tumšās robežlīnijas. Atdala šīs zonas un katras vielas sorbentu atsevišķi savāc 10 ml pudelē.
165.1.7. Atšķirīgi ekstrahē sorbentu, kas satur paraugu, un sorbentu, kurš satur standartmaisījumu: pievieno 2 ml metanola un, nepārtraukti maisot, ekstrahē vienu stundu. Izfiltrē maisījumu un atkārto ekstrakciju vēl 15 minūtes ar 2 ml metanola.
165.1.8. Apvieno ekstraktus un nakti tvaicē vakuuma eksikatorā, kas pildīts ar piemērotu žāvējošu vielu. Nesilda.
165.1.9. Sililē atlikumus (atbilstoši šā pielikuma 165.1.8.apakšpunktam saskaņā ar šā pielikuma 165.1.9.1. vai 165.1.9.2.apakšpunktu:
165.1.9.1. Ar mikrošļirci pievieno 200 ml BSTFA (atbilstoši šā pielikuma 163.9.1.apakšpunktam) un atstāj maisījumu slēgtā traukā uz 12 stundām istabas temperatūrā.
165.1.9.2. Ar mikrošļirci secīgi pievieno 200 ml HMDS (atbilstoši šā pielikuma 163.9.2.apakšpunktam) un 100 ml TMCS (atbilstoši šā pielikuma 163.9.3.apakšpunktam) un slēgtā traukā karsē maisījumu 30 minūtes 60 °C temperatūrā. Maisījumu atdzesē.
166. Gāzu hromatogrāfiju veic šādā kārtībā:
166.1. Hromatogrāfijas nosacījumi:
166.1.1. Kolonnai nodrošina izšķirtspēju R, kas ir vienāda ar 1,5 vai labāka, ja R = ((2d’(r2– r1))/(w1×w2)), kur
r1 un r2 – divu pīķu izdalīšanas laiki (minūtēs);
w1 un w2 – to pašu pīķu platumi pusaugstumā (mm);
d’ – pašrakstītāja ātrums (milimetros minūtē).
166.1.2. Par piemērotiem uzskata šādus kolonnas un gāzu hromatogrāfijas nosacījumus:
|
kolonna |
materiāls |
nerūsējošs tērauds |
|
garums |
200 cm |
|
|
iekšējais diametrs |
~ 3 mm |
|
|
pildījums |
10 % OV 17 uz Chromosorb WAW ar daļiņu izmēru 100–120 |
|
|
liesmas jonizācijas detektors |
||
|
temperatūras |
||
|
kolonna |
185 °C (izotermiska) |
|
|
detektors |
250 °C |
|
|
inžektors |
250 °C |
|
|
nesējgāze |
Slāpeklis |
|
|
plūsma |
45 ml/min |
|
166.1.3. Ūdeņraža un gaisa plūsmu ieregulē pēc ražotāja norādījumiem.
166.2. Ievada 1–3 ml šķīduma, ko iegūst gāzu hromatogrāfijā saskaņā ar šī pielikuma 165.1.9.apakšpunktu. Katru šķīdumu (atbilstoši šā pielikuma 165.1.9.apakšpunktam) ievada piecas reizes, izmēra pīķu laukumus, aprēķina vidējo un aprēķina pīķu laukumu attiecību: S = rezorcīna pīķu laukums/DHT pīķu laukums.
167. Rezorcīna koncentrāciju paraugā, kas izteikta masas procentos
(% m/m), aprēķina pēc formulas:
% rezorcīna = (4/M) × (Sparauga /Sstandartmaisījuma), kur
M – parauga daudzums (gramos) (atbilstoši šā pielikuma 165.1.1.apakšpunktam);
Sparauga – parauga šķīduma vidējā pīķu laukuma attiecība saskaņā ar šā pielikuma 166.2.2.apakšpunktu;
Sstandartmaisījuma – standartmaisījuma vidējā laukumu attiecība saskaņā ar šā pielikuma 166.2.2.apakšpunktu.
168. No viena parauga ar aptuveni 0,5 % rezorcīna saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,025 % vērtību4 .
13. Metanola kvantitatīva noteikšana attiecībā pret etanolu vai propān-2-olu
169. Ar metanola kvantitatīvas noteikšanas attiecībā pret etanolu vai propān-2-olu metodi gāzu hromatogrāfijā nosaka metanolu visu veidu kosmētikas līdzekļos (tostarp aerosolos). Var noteikt procentuālo koncentrāciju no 0 līdz 10 %.
170. Metanola saturu izsaka metanola masas procentos attiecībā pret etanolu vai propān-2-olu.
171. Kvantitatīvo noteikšanu veic gāzu hromatogrāfijā.
172. Metanola kvantitatīvas noteikšanas attiecībā pret etanolu vai propān-2-olu metodei izmanto šādus reaģentus1:
172.1. Metanols.
172.2. Absolūtais spirts.
172.3. Propān-2-ols.
172.4. Hloroforms, no kura ar ūdeni atmazgāti spirti.
173. Metanola kvantitatīvas noteikšanas attiecībā pret etanolu vai propān-2-olu metodei izmanto šādu iekārtu:
173.1. Gāzu hromatogrāfs ar katarometru aerosolu paraugiem, ar liesmas jonizācijas detektoru paraugiem, kas nav aerosoli.
173.2. Mērkolbas, 100 ml.
173.3. Pipetes, 2 ml, 20 ml, 0–1 ml.
173.4. Mikrošļirces 0–100 µl un 0–5 µl un (tikai aerosolu paraugiem) īpaša hermētiska šļirce ar ventili (5.attēls).
174. Metanola kvantitatīvu noteikšanu veic šādā kārtībā:
174.1. Parauga sagatavošana:
174.1.1. Aerosolu paraugus ņem saskaņā ar šī pielikuma 2.nodaļu un pēc tam analizē gāzu hromatogrāfijā ar šā pielikuma 174.2.1.apakšpunktā minētajiem nosacījumiem.
174.1.2. To kosmētikas līdzekļu paraugus, kuri nav aerosoli, ņem saskaņā ar šā pielikuma 2.nodaļu un atšķaida ar ūdeni, lai etanola vai propān-2-ola koncentrācija būtu 1–2 %, un pēc tam analizē gāzu hromatogrāfijā ar šā pielikuma 174.2.2.apakšpunktā minētajiem nosacījumiem.
174.2. Gāzu hromatogrāfija:
174.2.1. Aerosolu paraugu analīzei izmanto katarometru:
174.2.1.1. Kolonnu pilda ar 10 % Hallcomid M18 uz Chromosorb WAW ar daļiņu lielumu 100–200.
174.2.1.2. Kolonnai jānodrošina izšķirtspēja R, kas vienāda ar 1,5 vai labāka, ja R=((2) ((d’r2 – d’r1)/(w1 + w2))), kur
r1 un r2 – divu pīķu izdalīšanas laiki (minūtēs);
w1 un w2 – to pašu pīķu platumi pusaugstumā (mm);
d’ – pašrakstītāja ātrums (milimetros minūtē).
174.2.1.3. Šo izšķirtspēju ļauj nodrošināt šādi nosacījumi:
|
kolonna |
materiāls |
nerūsējošs tērauds |
|
garums |
3,5 m |
|
|
diametrs |
3 mm |
|
|
katarometra strāva |
150 mA |
|
|
nesējgāze |
hēlijs |
|
|
spiediens |
2,5 bar |
|
|
plūsma |
45 ml/min |
|
|
temperatūra |
||
|
inžektors |
||
|
detektors |
150 °C |
|
|
kolonnas kamera |
65 °C |
|
|
pīķu laukumu mērījumus var uzlabot, elektroniski integrējot |
||
174.2.2. Paraugiem, kas nav aerosoli:
174.2.2.1. Kolonnu pilda ar Chromosorb 105 vai Porapak QS un izmanto liesmas jonizācijas detektoru.
174.2.2.2. Kolonnai jānodrošina izšķirtspēja R, kas ir vienāda ar 1,5 vai labāka, ja R=((2) ((d’r2 – d’r1)/(w1 + w2))), kur
r1 un r2 – divu pīķu izdalīšanas laiki (minūtēs);
w1 un w2 – to pašu pīķu platumi pusaugstumā (mm);
d’ – pašrakstītāja ātrums (milimetros minūtē).
174.2.2.3. Šo izšķirtspēju nodrošina šādi nosacījumi:
|
kolonna |
materiāls |
nerūsējošs tērauds |
|
garums |
2 m |
|
|
diametrs |
3 mm |
|
|
elektrometra jutība |
8 × 10-10 A |
|
|
gāzes |
||
|
nesējgāze |
slāpeklis |
|
|
spiediens |
2,1 bar |
|
|
plūsma |
40 ml/min |
|
|
palīggāze |
ūdeņradis |
|
|
spiediens |
1,5 bar |
|
|
plūsma |
20 ml/min |
|
|
temperatūra |
||
|
inžektors |
150 °C |
|
|
detektors |
230 °C |
|
|
kolonnas kamera |
120–130 °C |
|
175. Standarta grafiku veido šādi:
175.1. Gāzu hromatogrāfijai saskaņā ar šī pielikuma 174.2.1.apakšpunktu (Hallcomid M18 kolonna) izmanto šādus standartmaisījumus (maisījumus sagatavo, mērot ar pipetēm, tomēr precīzo daudzumu nosaka, sverot pipeti vai kolbu pēc katras pievienošanas):
|
Procentuālā koncentrācija (m/m %) |
Metanols |
Etanols vai propān-2-ols (ml) |
Hloroforms, ko pievieno šādam tilpumam |
|
Aptuveni 2,5 % |
0,5 |
20 |
100 ml |
|
Aptuveni 5,0 % |
1,0 |
20 |
100 ml |
|
Aptuveni 7,5 % |
1,5 |
20 |
100 ml |
|
Aptuveni 10,0 % |
2,0 |
20 |
100 ml |
Saskaņā ar šā pielikuma 174.2.1.apakšpunktu ievada hromatogrāfā 2–3 μl. Aprēķina katra maisījuma pīķu laukumu attiecību (metanols/etanols) vai (metanols/propān-2-ols). Uzzīmē standarta grafiku, kur uz x ass % atliek metanola attiecību pret etanolu vai propān-2-olu, uz y ass atliek pīķu laukumu attiecību (metanols/etanols) vai (metanols/propān-2-ols).
175.2. Gāzu hromatogrāfijai saskaņā ar šā pielikuma 174.2.2.apakšpunktu (Porapak QS vai Chromosorb 105) izmanto šādus standartmaisījumus (maisījumus sagatavo, mērot ar mikrošļirci un pipeti, tomēr precīzo daudzumu nosaka, sverot pipeti vai kolbu tūlīt pēc katras pievienošanas):
|
Procentuālā koncentrācija (m/m %) |
Metanols |
Etanols vai propān-2-ols (ml) |
Ūdens, ko pievieno šādam tilpumam |
|
Aptuveni 2,5 % |
50 |
2 |
100 ml |
|
Aptuveni 5,0 % |
100 |
2 |
100 ml |
|
Aptuveni 7,5 % |
150 |
2 |
100 ml |
|
Aptuveni 10,0 % |
200 |
2 |
100 ml |
Saskaņā ar šā pielikuma 174.2.2.apakšpunktu ievada hromatografā 2–3 μl. Aprēķina katra maisījuma pīķu laukumu attiecību (metanols/etanols) vai (metanols/propān-2-ols). Uzzīmē standarta grafiku, kur uz x ass % atliek metanola attiecību pret etanolu vai propān-2-olu, uz y ass atliek pīķu laukumu attiecību (metanols/etanols) vai (metanols/propān-2-ols).
175.3. Standarta grafikam jābūt taisnei.
176. No viena parauga ar aptuveni 5 % metanola saturu attiecībā pret etanolu vai propān-2-olu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,25 %4.
14. Dihlormetāna un 1,1,1-trihlormetāna noteikšana
177. Ar dihlormetāna un 1,1,1-trihlormetāna noteikšanas metodi nosaka dihlormetānu (metilēnhlorīdu) un 1,1,1-trihloretānu (metilhloroformu) visos kosmētikas līdzekļos, kuros tas varētu būt.
178. Dihlormetāna un 1,1,1-trihloretāna saturu, ko paraugā nosaka saskaņā ar dihlormetāna un 1,1,1-trihlormetāna noteikšanas metodi, izsaka masas procentos.
179. Pielietojot dihlormetāna un 1,1,1-trihlormetāna noteikšanas metodi, izmanto gāzu hromatogrāfiju ar hloroformu kā iekšējo standartu.
180. Dihlormetāna un 1,1,1-trihlormetāna noteikšanas metodei izmanto šādus reaģentus1:
180.1. Hloroforms (CHCl3).
180.2. Tetrahlorogleklis (CCl4).
180.3. Dihlormetāns (CH2Cl2).
180.4. 1,1,1-trihloretāns (CH3CCl3).
180.5. Acetons.
180.6. Slāpeklis.
181. Dihlormetāna un 1,1,1-trihlormetāna noteikšanas metodei izmanto šādu aprīkojumu:
181.1. Standarta laboratorijas aprīkojums.
181.2. Gāzu hromatogrāfs, kas aprīkots ar katarometru.
181.3. Pudele šķīduma pārnešanai, 50–100 ml (atbilstoši šā pielikuma 19.punktam).
181.4. Gāzes spiedšļirce, 25 vai 50 μl (atbilstoši šā pielikuma 19.2.2.apakšpunktam).
182. Dihlormetāna un 1,1,1-trihlormetāna noteikšanu veic šādā kārtībā:
182.1. Spiedienam nepakļauts paraugs: precīzi iesver paraugu koniskā kolbā ar aizbāzni. Ievada precīzi nosvērtu daudzumu, kas ir vienāds ar iepriekš pieņemto dihlormetāna un 1,1,1-trihloretāna daudzumu paraugā hloroforma, kurš ir iekšējais standarts. Rūpīgi sajauc.
182.2. Paaugstinātam spiedienam pakļauts paraugs: izmanto metodi, kas aprakstīta šā pielikuma 2.nodaļā, bet ar šādiem precizējumiem:
182.2.1. Kad paraugs ir pārnests uz pudeli šķīduma pārnešanai, tajā ievada tādu hloroforma (kas ir iekšējais standarts) daudzumu, kas vienāds ar iepriekš pieņemto dihlormetāna un (vai) 1,1,1-trihloretāna daudzumu paraugā. Rūpīgi sajauc. Tukšu ventili izskalo ar 0,5 ml tetrahloroglekļa. Pēc žāvēšanas, pamatojoties uz starpību, precīzi nosaka iekšējā standarta pievienoto masu.
182.2.2. Kad šļirce ir piepildīta ar paraugu, tās uzgali iztīra ar slāpekli, lai pirms ievadīšanas hromatogrāfā nepaliktu atlikumi.
182.2.3. Pēc katra parauga paņemšanas ventiļa virsmu un pārnešanas ierīci vairākas reizes izskalo ar acetonu (ja nepieciešams, lietojot injekciju šļirci) un pēc tam rūpīgi nožāvē ar slāpekli.
182.2.4. Katrai analīzei veic mērījumus ar divām dažādām pudelēm šķīduma pārnešanai, piecus mērījumus ar katru pudeli.
183. Dihlormetāna un 1,1,1-trihlormetāna noteikšanas hromatogrāfijas apstākļi:
183.1. Pirmskolonnas nosacījumi ir šādi:
183.1.1. Caurule: nerūsējošs tērauds.
183.1.2. Garums: 300 mm.
183.1.3. Diametrs: 3 vai 6 mm.
183.1.4. Pildījums: tā pati viela, ar ko pilda analītisko kolonnu.
183.2. Kolonnas nekustīgo fāzi veido Hallcomid M 18 uz hromosorba. Kolonna nodrošina izšķirtspēju R, kas ir vienāda ar 1,5 vai labāka, ja:
![]() , kur
, kur
r1 un r2 – izdalīšanas laiki (minūtēs);
W1 un W2 – pīķu platumi pusaugstumā (milimetros);
d' – pašrakstītāja ātrums (milimetros minūtē).
183.3. Piemērs. Šādas kolonnas dod vēlamos rezultātus:
|
Kolonna |
I |
II |
|
Materiāls |
nerūsējoša tērauda caurule |
nerūsējoša tērauda caurule |
|
Garums |
350 cm |
400 cm |
|
Diametrs |
3 mm |
6 mm |
|
Nesējs: |
||
|
hromosorbs |
WAW |
WAW-DMCS-HP |
|
sieta analīze |
daļiņu lielums 100–120 |
daļiņu lielums 60–80 |
|
Nekustīgā fāze |
Hallcomid M 18, 10 % |
Hallcomid M 18, 20 % |
Temperatūra var atšķirties atkarībā no iekārtas funkcijas. Šajos piemēros tā ir noteikta šāda:
|
Kolonna |
I |
II |
|
Temperatūra: |
||
|
kolonna |
65 °C |
75 °C |
|
inžektors |
150 °C |
125 °C |
|
detektors |
150 °C |
200 °C |
|
Nesējgāze: |
||
|
hēlija plūsmas ātrums |
45 ml/min |
60 ml/min |
|
spiediens inžektorā |
2,5 bar |
2 bar |
|
iesmidzināšana |
15 μl |
15 μl |
184. Lai noteiktu signāla attiecību pret masas vienību, koniskā kolbā ar aizbāzni sagatavo šādu precīzi nosvērtu maisījumu: dihlormetāns 30 % (m/m), 1,1,1-trihloretāns 35 % (m/m), hloroforms 35 % (m/m).
185. Aprēķinus veic šādā kārtībā:
185.1. Signāla attiecības pret masas vienību vielai p aprēķins attiecībā pret vielu a, ko izmanto kā iekšējo standartu. Ja pirmā viela ir p, kur
kp – tās signāla attiecība pret masas vienību;
mp – tās masa maisījumā;
Ap – tās pīķa laukums;
otrā viela ir a, kur
ka – tās signāla attiecība pret masas vienību;
Ma – tās masa maisījumā;
Aa – tās pīķa laukums, tad:
![]() .
.
Piemēram, ir iegūtas šādas signāla attiecības pret masas vienību (hloroformam: k = 1):
dihlormetānam: k1 = 0,78±0,03
1,1,1-trihloretānam: k1 = 0,78±0,03.
185.2. Aprēķina % (m/m) dihlormetāna un 1,1,1-trihloretāna analizējamā paraugā. Ja:
ma – ievadītā hloroforma masa (gramos);
Ms – analizējamā parauga masa (gramos);
Aa – hloroforma pīķa laukums;
A1 – dihlormetāna pīķa laukums;
A2 – 1,1,1-trihloretāna pīķa laukums, tad:
![]()
![]()
186. No viena parauga ar dihlormetāna un (vai) 1,1,1-trihloretāna saturu 25 % (m/m) divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 2,5 % (m/m)4.
15. Hinolīn-8-ola un bis(8-hidroksihinolīn)sulfāta pierādīšana un noteikšana
187. Ar hinolīn-8-ola un bis(8-hidroksihinolīn)sulfāta pierādīšanas un noteikšanas metodi pierāda un nosaka hinolīn-8-olu un tā sulfātu.
188. Noteiktā hinolīn-8-ola un bis(8-hidroksihinolīn) sulfāta saturu izsaka hinolīn-8-ola masas procentos.
189. Hinolīn-8-ola un bis(8-hidroksihinolīn) sulfātu pierāda, veicot plānslāņa hromatogrāfiju, un nosaka, veicot tāda kompleksa spektrofotometriju 410 nm, kas iegūts reakcijā ar Fēlinga šķīdumu.
190. Hinolīn-8-ola un bis(8-hidroksihinolīn)sulfāta pierādīšanas un noteikšanas metodei izmanto šādus reaģentus1:
190.1. Hinolīn-8-ols.
190.2. Benzols. Tā kā benzols ir toksisks, ar to jāstrādā īpaši piesardzīgi.
190.3. Hloroforms.
190.4. Nātrija hidroksīda ūdens šķīdums, 50 % (m/m).
190.5. Vara sulfāta pentahidrāts.
190.6. Kālija nātrija tartrāts.
190.7. 1 M sālsskābe.
190.8. 0,5 M sērskābe.
190.9. Nātrija hidroksīds, 1 M šķīdums.
190.10. Etanols.
190.11. Butān-1-ols.
190.12. Ledus etiķskābe.
190.13. 0,1 n sālsskābe.
190.14. Celīts 545 vai līdzvērtīgs.
190.15. Standartšķīdumi:
190.15.1. Iesver 100 mg hinolīn-8-ola 100 ml mērkolbā. Izšķīdina nedaudz sērskābes (atbilstoši šā pielikuma 190.8.apakšpunktam). Uzpilda līdz zīmei ar sērskābi.
190.15.2. Iesver 100 mg hinolīn-8-ola 100 ml mērkolbā. Izšķīdina etanolā. Uzpilda līdz zīmei ar etanolu un sajauc.
190.16. Fēlinga šķīdums.
Šķīdums A: iesver 7 g vara sulfāta pentahidrāta 100 ml mērkolbā. Izšķīdina nelielā daudzumā ūdens. Uzpilda līdz zīmei ar ūdeni un sajauc.
Šķīdums B: iesver 35 g kālija nātrija tartrāta 100 ml mērkolbā. Izšķīdina 50 ml ūdens. Pievieno 20 ml nātrija hidroksīda (atbilstoši šā pielikuma 190.4.apakšpunktam). Uzpilda līdz zīmei ar ūdeni un sajauc. Tieši pirms lietošanas ar pipeti iepilina 10 ml šķīduma A un 10 ml šķīduma B 100 ml mērkolbā. Uzpilda līdz zīmei un sajauc.
190.17. Eluenti plānslāņa hromatogrāfijai:
190.17.1. Butān-1-ols/etiķskābe/ūdens 80:20:20 (v/v/v).
190.17.2. Hloroforms/etiķskābe 95:5 (v/v).
190.18. 2,6-dihlor-4-(hlorimino)cikloheksa-2,5-dienons, 1 % (m/v) šķīdums etanolā.
190.19. Nātrija karbonāts, 1 % (m/v) šķīdums ūdenī.
190.20. Etanols, 30 % (v/v) šķīdums ūdenī.
190.21. Dinātrija dihidrogēnetilēndiamīntetraacetāts, 5 % (m/v) šķīdums ūdenī.
190.22. Buferšķīdums, pH 7: iesver 27g bezūdens kālija dihidrogēnortofosfāta un 70 g dikālija hidrogēnortofosfāta trihidrāta viena litra mērkolbā. Uzpilda līdz zīmei ar ūdeni.
190.23. Lietošanai gatavas plānslāņa plates, kuru biezums ir 0,25 mm (tai skaitā Merck Kieselgel 60 vai līdzvērtīgas), pirms lietošanas apsmidzina ar 10 ml reaģenta (atbilstoši šā pielikuma 190.21.apakšpunktam) un nožāvē 80 °C.
191. Hinolīn-8-ola un bis(8-hidroksihinolīn)sulfāta pierādīšanas un noteikšanas metodei izmanto šādu aprīkojumu:
191.1. Apaļkolba ar pieslīpētu kaklu, 100 ml.
191.2. Mērkolbas.
191.3. Mērpipetes, 10 un 5 ml.
191.4. pipetes, 20, 15, 10 un 5 ml.
191.5. Dalāmās piltuves, 100, 50 un 25 ml.
191.6. Kroku filtrpapīrs ar 90 mm diametru.
191.7. Rotācijas ietvaicētājs.
191.8. Atteces dzesinātājs ar pieslīpētu kaklu.
191.9. Spektrofotometrs.
191.10. Kivetes ar 10 mm optiskā ceļa garumu.
191.11. Maisītājs ar elektrisko apsildi.
191.12. Stikla hromatogrāfijas kolonnas izmēri: 160 mm gara ar 8 mm diametru, sašaurināta apakšgalā, kur ir stiklšķiedras aizbāznis un adapters augšgalā spiediena radīšanai.
192. Hinolīn-8-ola un bis(8-hidroksihinolīn)sulfāta pierādīšanu veic šādā kārtībā:
192.1. Šķidrie paraugi:
192.1.1. Analizējamā parauga daļas pH noregulē uz 7,5 un 10 μl uznes uz iepriekš apstrādātas silikagela plānslāņa plates (atbilstoši šā pielikuma 190.23.apakšpunktam) starta līnijas.
192.1.2. Standartšķīduma (atbilstoši šā pielikuma 190.15.2.apakšpunktam) 10 un 30 μl uznes vēl divos starta līnijas punktos, pēc tam plati attīsta vienā no diviem eluentiem (atbilstoši šā pielikuma 190.17.apakšpunktam).
192.1.3. Kad šķīdinātāja fronte ir pavirzījusies par 150 mm, plati žāvē 110 °C (15 minūtes). Ultravioletā gaismā (366 nm) inolīn-8-ola plankumi fluorescē dzelteni.
192.1.4. Apsmidzina plati ar nātrija karbonāta šķīdumu (atbilstoši šā pielikuma 190.19.apakšpunktam). Nožāvē un apsmidzina ar 2,6-dihlor-4-(hlorimino)cikloheksa-2,5-dienona šķīdumu (atbilstoši šā pielikuma 190.18.apakšpunktam). Hinolīn-8-ols kļūst redzams kā zils plankums.
192.2. Cietie/sausie paraugi vai krēmi:
192.2.1. Disperģē 1 g parauga 5 ml buferšķīduma (atbilstoši šā pielikuma 190.22.apakšpunktam). Pēc tam pārnes 10 ml hloroforma uz dalāmo piltuvi un sakrata. Pēc hloroforma slāņa atdalīšanas ūdens slāni ekstrahē vēl divas reizes ar 10 ml hloroforma. Apvienotos un izfiltrētos hloroforma ekstraktus gandrīz pilnīgi ietvaicē 10 ml apaļkolbā ar rotācijas ietvaicētāju. Izšķīdina atlikumu 2 ml hloroforma un 10 un 30 μl iegūtā šķīduma uznes uz silikagela plānslāņa plates (atbilstoši šā pielikuma 190.23.apakšpunktam) saskaņā ar šā pielikuma 192.1.apakšpunktā minēto metodi.
192.2.2. Uz plates uznes 10 un 30 μl standartšķīduma (atbilstoši šā pielikuma 190.15.2.apakšpunktam) un turpina saskaņā ar šā pielikuma 192.1.2., 192.1.3. un 192.1.4.apakšpunktu.
193. Hinolīn-8-ola un bis(8-hidroksihinolīn)sulfāta noteikšanu veic šādā kārtībā:
193.1. Šķidrie paraugi:
193.1.1. Iesver 5 g parauga 100 ml apaļkolbā. Pievieno 1 ml sērskābes šķīduma (atbilstoši šā pielikuma 190.8.apakšpunktam) un maisījumu ietvaicē līdz gandrīz sausam stāvoklim 50 °C pazeminātā spiedienā.
193.1.2. Izšķīdina atlikumu 20 ml silta ūdens. Pārnes uz 100 ml mērkolbu. Skalo trīs reizes ar 20 ml ūdens. Uzpilda līdz 100 ml ar ūdeni un sajauc.
193.1.3. Iepilina 5 ml šī šķīduma 50 ml dalāmajā piltuvē. Pievieno 10 ml Fēlinga šķīduma (atbilstoši šā pielikuma 190.16.apakšpunktam). Ekstrahē hinolīn-8-ola vara kompleksu [vara oksīnu (ISO)], ko iegūst ar trīsreiz 8 ml hloroforma.
193.1.4. Izfiltrē un savāc hloroforma slāņus 25 ml mērkolbā. Uzpilda līdz zīmei ar hloroformu un sakrata. Nosaka dzeltenā šķīduma optisko blīvumu attiecībā pret hloroformu 410 nm.
193.2. Cietie/sausie paraugi vai krēmi:
193.2.1. Iesver 0,500 g parauga 100 ml apaļkolbā (atbilstoši šā pielikuma 191.1.apakšpunktam). Pievieno 30 ml benzola un 20 ml sālsskābes (atbilstoši šā pielikuma 190.7.apakšpunktam). Izmantojot atteces dzesinātāju, maisot vāra kolbas saturu 30 minūtes.
193.2.2. Kolbas saturu pārnes uz 100 ml dalāmo piltuvi. Skalo ar 5 ml 1 N HCl. Pārnes ūdens fāzi uz apaļkolbu un mazgā benzola fāzi ar 5 ml sālsskābes.
193.2.3. Ja turpmāko apstrādi traucē emulsijas, 0,500 g parauga sajauc ar 2 g celīta545 (atbilstoši šā pielikuma 190.14.apakšpunktam), lai veidojas brīvi plūstošs pulveris. Maisījumu mazās devās pārnes uz stikla hromatogrāfijas kolonnu (atbilstoši šā pielikuma 191.12.apakšpunktam). Pēc katras pievienošanas sablīvē kolonnas pildījumu. Tiklīdz viss maisījums ir pārnests uz kolonnu, eluē ar sālsskābi (atbilstoši šā pielikuma 190.13.apakšpunktam) tā, lai apmēram 10 minūtēs iegūst 10 ml eluāta (ja nepieciešams, var eluēt nelielā slāpekļa spiedienā). Eluējot jānodrošina, lai virs kolonnas pildījuma nepārtraukti ir nedaudz sālsskābes. Pirmos 10 ml eluāta turpina apstrādāt saskaņā ar šā pielikuma 193.2.4.apakšpunktu.
193.2.4. Savāktās ūdens fāzes (atbilstoši šā pielikuma 193.2.2.apakšpunktam) vai eluātu (atbilstoši šā pielikuma 193.2.3.apakšpunktam) ar rotācijas ietvaicētāju pazeminātā spiedienā ietvaicē līdz gandrīz sausam stāvoklim.
193.2.5. Atlikumu izšķīdina 6 ml nātrija hidroksīda šķīduma (atbilstoši šā pielikuma 190.9.apakšpunktam). Pievieno 20 ml Fēlinga šķīduma (atbilstoši šā pielikuma 190.16.apakšpunktam) un kolbas saturu pārnes uz 50 ml dalāmo piltuvi. Kolbu izskalo ar 8 ml hloroforma. Sakrata un izfiltrē hloroforma fāzi 50 ml mērkolbā.
193.2.6. Atkārto ekstrakciju trīs reizes ar 8 ml hloroforma. Izfiltrē hloroforma fāzes un savāc 50 ml kolbā. Uzpilda līdz zīmei ar hloroformu un sakrata. Izmēra dzeltenā šķīduma optisko blīvumu attiecībā pret hloroformu 410 nm.
194. Kalibrēšanas līkni veido šādi: četrās 100 ml apaļkolbās, no kurām katrā ir 3 ml 30 % etanola ūdens šķīduma (atbilstoši šā pielikuma 190.20.apakšpunktam), ar pipeti iepilina 5, 10, 15 un 20 ml devu standartšķīduma (atbilstoši šā pielikuma 190.15.1.apakšpunktam), kas atbilst 5, 10, 15 un 20 mg hinolīn-8-ola. Pēc tam rīkojas saskaņā ar šā pielikuma 193.1.apakšpunktu.
195. Aprēķinu veic šādā kārtībā:
195.1. Šķidrie paraugi:
Hinolīn-8-ola saturs (% m/m) = ![]() , kur
, kur
A – miligrami hinolīn-8-ola uz kalibrēšanas līknes (atbilstoši šā pielikuma 194.punktam);
m – noteikšanai ņemtā parauga (atbilstoši šā pielikuma 193.1.1.apakšpunktam) masa (miligramos).
195.2. Cietie/sausie paraugi vai krēmi:
Hinolīn-8-ola saturs (% (m/m)) = ![]() , kur
, kur
a – miligrami hinolīn-8-ola uz kalibrēšanas līknes (atbilstoši šā pielikuma 194.punktam);
m – noteikšanai ņemtā parauga (atbilstoši šā pielikuma 193.2.1.apakšpunktam) masa (miligramos).
196. Ja hinolīn-8-ola saturs ir aptuveni 0,3 %, no tā paša parauga divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,02 % vērtību4.
16. Amonjaka noteikšana
197. Ar amonjaka noteikšanas metodi nosaka brīvo amonjaku kosmētikas līdzekļos.
198. Ar amonjaka noteikšanas metodi noteikto amonjaka saturu paraugā izsaka amonjaka masas procentos.
199. Analizējamam kosmētikas līdzekļa parauga daudzumam, kas izšķīdināts metanola–ūdens šķīdumā, pievieno bārija hlorīda šķīdumu. Filtrējot vai centrifugējot atdala visas nogulsnes. Šajā procedūrā, destilējot ar ūdens tvaiku, bez zudumiem saglabā amonjaku dažos amonija sāļos, piemēram, hidrogēnkarbonātā un taukskābēs, izņemot amonija acetātu. Amonjaku ar ūdens tvaiku destilē no filtrāta vai centrifugāta un nosaka, potenciometriski vai citādi titrējot.
200. Amonjaka noteikšanas metodei izmanto šādus reaģentus1:
200.1. Metanols.
200.2. Bārija hlorīda dihidrāts, 25 % (m/v) šķīdums.
200.3. Ortoborskābe, 4 % (m/v) šķīdums.
200.4. Sērskābe, 0,25 M standartšķīdums.
200.5. Pretputu līdzeklis.
200.6. Nātrija hidroksīds, 0,5 M standartšķīdums.
200.7. Indikators (ja nepieciešams): sajauc 5 ml 0,1 % (m/v) metilsarkanā šķīduma etanolā ar 2 ml 0,1 % (m/v) metilēnzilā šķīduma ūdenī.
201. Amonjaka noteikšanas metodei izmanto šādu aprīkojumu:
201.1. Standarta laboratorijas aprīkojums.
201.2. Centrifūga ar 100 ml pudelēm, kam ir aizbāžņi.
201.3. Iekārta destilēšanai ar ūdens tvaiku.
201.4. Potenciometrs.
201.5. Indikācijas stikla elektrods un dzīvsudraba(I) hlorīda (kalomela) standartelektrods.
202. Amonjaka noteikšanu veic šādā kārtībā:
202.1. Iesver 100 ml mērkolbā paraugu ar masu (m), kas atbilst 150 mg amonjaka maksimumam.
202.2. Pievieno 10 ml ūdens, 10 ml metanola un 10 ml bārija hlorīda šķīduma (atbilstoši šā pielikuma 200.2.apakšpunktam). Uzpilda ar metanolu līdz 100 ml.
202.3. Sajauc un uz nakti atstāj ledusskapī (5 °C).
202.4. Pēc tam filtrē vai 10 minūtes slēgtās mēģenēs centrifugē vēl aukstu šķīdumu, lai iegūtu dzidru filtrātu vai centrifugātu.
202.5. Ar pipeti iepilina 40 ml dzidrā šķīduma iekārtā destilēšanai ar ūdens tvaiku (atbilstoši šā pielikuma 201.3.apakšpunktam), pēc tam, ja nepieciešams, 0,5 ml pretputu līdzekļa.
202.6. Destilē un savāc 200 ml destilāta 250 ml vārglāzē, kurā ir 10 ml sērskābes standartšķīduma (atbilstoši šā pielikuma 200.4.apakšpunktam) un 0,1 ml indikatora (atbilstoši šā pielikuma 200.7.apakšpunktam).
202.7. Attitrē skābes pārākumu ar nātrija hidroksīda standartšķīdumu (atbilstoši šā pielikuma 200.6.apakšpunktam).
202.8. Potenciometriskajai noteikšanai savāc 200 ml destilāta 250 ml vārglāzē, kurā ir 25 ml ortoborskābes šķīduma (atbilstoši šā pielikuma 200.3.apakšpunktam), un titrē ar sērskābes standartšķīdumu (atbilstoši šā pielikuma 200.4.apakšpunktam), reģistrējot neitralizācijas līkni.
203. Aprēķinus veic šādā kārtībā:
203.1. Aprēķins attitrējot. Ja:
V1 – izlietotā nātrija hidroksīda šķīduma tilpums (mililitros);
M1 – tā faktiskā molaritāte (atbilstoši šā pielikuma 200.6.apakšpunktam);
M2 – sērskābes šķīduma (atbilstoši šā pielikuma 200.4.apakšpunktam) faktiskais titrs;
m – noteikšanai ņemtā parauga (atbilstoši šā pielikuma 202.1.apakšpunktam) masa (miligramos), tad:
amonjaka % (m/m) = ![]()
203.2. Aprēķins, ja veic tiešu potenciometrisku titrēšanu. Ja:
V2 – izlietotā sērskābes šķīduma tilpums (mililitros);
M2 – tā faktiskā molaritāte (atbilstoši šā pielikuma 200.4.apakšpunktam);
M – noteikšanai ņemtā parauga (atbilstoši šā pielikuma 202.1.apakšpunktam) masa (miligramos), tad:
amonjaka % (m/m) = ![]()
204. Ja amonjaka saturs ir aptuveni 6 %, viena parauga divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,6 % vērtību4.
17. Nitrometāna pierādīšana un noteikšana
205. Ar nitrometāna pierādīšanas un noteikšanas metodi pierāda un nosaka nitrometānu, ja tā saturs ir līdz 0,3 %, aerosolveida kosmētikas līdzekļos.
206. Nitrometāna saturu paraugā izsaka amonjaka masas procentos visā aerosola balonā.
207. Nitrometānu pierāda ar krāsu reakciju. Nitrometānu nosaka gāzu hromatogrāfijā pēc iekšējā standarta pievienošanas.
17.1. Nitrometāna pierādīšana
208. Nitrometāna pierādīšanai izmanto šādus reaģentus1:
208.1. Nātrija hidroksīds, 0,5 M šķīdums.
208.2. Folina reaģents: izšķīdina 0,1 g nātrija 3,4-dihidro-3,4-dioksonaftalīn-1-sulfonātu ūdenī un atšķaida līdz 100 ml.
209. Nitrometāna pierādīšanu veic pievienojot 1 ml parauga 10 ml šā pielikuma 208.1.apakšpunktā un 1 ml šā pielikuma 208.2.apakšpunktā minētās vielas. Violets krāsojums liecina par nitrometāna klātbūtni.
17.2. Nitrometāna noteikšana
210. Nitrometāna noteikšanai izmanto šādus reaģentus1:
210.1. Hloroforms (iekšējais standarts 1).
210.2. 2,4-dimetilheptāns (iekšējais standarts 2).
210.3. Etanols, 95 %.
210.4. Nitrometāns.
210.5. Hloroforma standartšķīdums: nosvērtā 25 ml mērkolbā ievada apmēram 650 mg hloroforma. Vēlreiz precīzi nosver kolbu un tās saturu. Uzpilda līdz 25 ml ar 95 % etanolu. Nosver un aprēķina hloroforma masas procentus šajā šķīdumā.
210.6. 2,4-dimetilheptāna standartšķīdums: hloroforma standartšķīdumu uzpilda saskaņā ar šā pielikuma 210.5.apakšpunktu, bet 25 ml mērkolbā iesver 270 mg 2,4-dimetilheptāna.
211. Nitrometāna noteikšanai izmanto šādu aprīkojumu:
211.1. Gāzu hromatogrāfs ar liesmas jonizācijas detektoru.
211.2. Iekārta paraugu ņemšanai no aerosoliem (pudele šķīduma pārnešanai, mikrošļirces savienotāji utt.) saskaņā ar šī pielikuma 2.nodaļu.
211.3. Laboratorijas standartaprīkojums.
212. Nitrometāna noteikšanu veic šādā kārtībā:
212.1. Parauga sagatavošana: nosvērtā 100 ml šķīduma pārnešanas pudelē, kas izskalota vai iztukšota saskaņā ar šī pielikuma 17., 18. un 19.punktā minēto procedūru, ievada apmēram 5 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 210.5. vai 210.6.apakšpunktam). Lieto 10 vai 20 ml stikla šļirci bez adatas, kas pielāgota pārnešanas ierīcei saskaņā ar šā pielikuma 17., 18. un 19.punktā minēto paņēmienu. Vēlreiz nosver, lai noteiktu ievadīto daudzumu. Ar tādu pašu paņēmienu uz šo pudeli pārnes apmēram 50 g parauga, kas ņemts no aerosola balona. Vēlreiz nosver, lai noteiktu pārnestā parauga daudzumu. Labi sajauc. Ar minēto mikrošļirci (atbilstoši šā pielikuma 211.2.apakšpunktam) ievada apmēram 10 μl. Ievada piecas reizes.
212.2. Standarta gatavošana: precīzi iesver 50 ml mērkolbā apmēram 500 mg nitrometāna un 500 mg hloroforma, vai 210 mg 2,4-dimetilheptāna. Uzpilda līdz zīmei ar 95 % etanolu. Rūpīgi sajauc. Pārnes 5 ml šī šķīduma uz 20 ml mērkolbu. Uzpilda līdz zīmei ar 95 % etanolu. Ar minēto mikrošļirci (atbilstoši šā pielikuma 210.2.apakšpunktam) ievada apmēram 10 μl. Ievada piecas reizes.
212.3. Gāzu hromatogrāfijas nosacījumi:
212.3.1. Kolonnai ir divas daļas: pirmā ar didecilftalātu ar Gas Chrom Q pildījumu, otrā ar Ucon 50 HB 280X ar Gas Chrom Qpildījumu. Sagatavotajai kombinētajai kolonnai jānodrošina izšķirtspēja R, kas ir vienāda ar 1,5 vai labāka:
![]() , kur
, kur
r1 un r2 – izdalīšanas laiki (minūtēs);
W1 un W2 – pīķu platumi pusaugstumā (milimetros);
d' – pašrakstītāja ātrums (milimetros minūtē).
Piemēram, šīs divas daļas nodrošina vajadzīgo izšķirtspēju:
Kolonna A
Materiāls: nerūsējošs tērauds.
Garums: 1,5 m.
Diametrs: 3 mm.
Pildījums: 20 % didecilftalāts uz Gas Chrom Q (daļiņu lielums 100–120).
Kolonna B
Materiāls: nerūsējošs tērauds.
Garums: 1,5 m.
Diametrs: 3 mm.
Pildījums: 20 % Ucon 50 HB 280X uz GasChrom Q (daļiņu lielums 100–120).
212.3.2. Piemērots liesmas jonizācijas detektora elektrometra jutības uzstādījums ir 8 × 10-10 A.
212.3.3. Temperatūra:
212.3.3.1. Inžektorā: 150 °C.
212.3.3.2. Detektorā: 150 °C.
212.3.3.3. Kolonnā: 50–80 °C atkarībā no attiecīgās kolonnas un iekārtas.
212.3.4. Piemērotā gāze:
212.3.4.1. Nesējgāze: slāpeklis.
212.3.4.2. Spiediens: 2,1 bar.
212.3.4.3. Plūsma: 40 ml/min.
212.3.4.4. Pievade detektoram: pēc detektora izgatavotāju norādījumiem.
213. Aprēķinus veic šādā kārtībā:
213.1. Nitrometāna signāla attiecība pret masas vienību, ko aprēķina attiecībā pret izmantoto iekšējo standartu. Ja nitrometānu apzīmē ar n, tad:
kn – tā signāla attiecība pret masas vienību;
m'n – tā masa (gramos) maisījumā;
S'n – tā pīķa laukums.
Ja iekšējo standartu, hloroformu vai 2,4-dimetilheptānu, apzīmē ar c, tad:
m'c – tā masa (gramos) maisījumā;
S'c – tā pīķa laukums, un:
![]() (kn ir iekārtas
funkcija)
(kn ir iekārtas
funkcija)
213.2. Nitrometāna koncentrācija paraugā, ja nitrometānu apzīmē ar n, tad:
kn – tā signāla attiecība pret masas vienību;
Sn – tā pīķa laukums.
Ja iekšējo standartu, hloroformu vai 2,4-dimetilheptānu, apzīmē ar c, tad:
mc – tā masa (gramos) maisījumā;
Sc – tā pīķa laukums;
M – pārnestā aerosola masa (gramos), un
nitrometāna % (m/m) paraugā ir: ![]()
214. Ja nitrometāna saturs ir aptuveni 0,3 % (m/m), no viena parauga divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,03 % (m/m) vērtību4.
18. Merkaptoetiķskābes pierādīšana un noteikšana ilgviļņu, matu iztaisnošanas un epilācijas līdzekļos
215. Ar merkaptoetiķskābes pierādīšanas un noteikšanas metodi pierāda un nosaka merkaptoetiķskābi ilgviļņu, matu iztaisnošanas un epilācijas līdzekļos, kuros var būt citi reducējoši aģenti.
216. Merkaptoetiķskābes saturu paraugā izsaka merkaptoetiķskābes masas procentos.
217. Merkaptoetiķskābi pierāda ar krāsu reakciju, veicot plānslāņa hromatogrāfiju, un nosaka jodometriski vai ar gāzu hromatogrāfijas palīdzību.
18.1. Merkaptoetiķskābes pierādīšana ar krāsu reakciju
218. Merkaptoetiķskābes pierādīšanai ar krāsu reakciju izmanto šādus reaģentus1:
218.1. Svina acetāta papīrs.
218.2. Sālsskābes šķīdums (viena tilpuma vienība koncentrētas sālsskābes un viena tilpuma vienība ūdens).
219. Merkaptoetiķskābes pierādīšanu ar krāsu reakciju veic šādā kārtībā:
219.1. Merkaptoetiķskābes pierādīšana krāsu reakcijā ar svina acetātu: vienu pilienu analizējamā parauga liek uz svina acetāta papīra. Ja tas nokrāsojas spilgti dzeltens, tad, iespējams, tajā ir merkaptoetiķskābe. Jutība 0,5 %.
219.2. Neorganisko sulfīdu raksturojums pēc hidrogēnsulfīda veidošanās skābinot: mēģenē ievada dažus miligramus analizējamā parauga. Pievieno 2 ml destilēta ūdens un 1 ml sālsskābes (atbilstoši šā pielikuma 218.2.apakšpunktam). Rodas hidrogēnsulfīds, ko konstatē pēc smaržas, un uz svina acetāta papīra veidojas melnas svina sulfīda nogulsnes. Jutība 50 ppm.
219.3. Sulfītu raksturojums pēc sēra dioksīda veidošanās skābinot: rīkojas saskaņā ar šā pielikuma 219.2.apakšpunktu. Uzvāra. Sēra dioksīdu konstatē pēc smaržas un tā reducējošajām īpašībām, piemēram, attiecībā pret permanganāta joniem.
18.2. Merkaptoetiķskābes pierādīšana plānslāņa hromatogrāfijā
220. Merkaptoetiķskābes pierādīšanai plānslāņa hromatogrāfijā izmanto šādus reaģentus1:
220.1. Merkaptoetiķskābe (tioglikolskābe), tīrība vismaz 98 %, ko pārbauda jodometriski.
220.2. 2,2'ditiodi(etiķskābe), tīrība vismaz 99 %, ko pārbauda jodometriski.
220.3. 2-merkaptopropionskābe (tiopienskābe), vismaz 95 %, ko pārbauda jodometriski.
220.4. 3-merkaptopropionskābe, tīrība vismaz 98 %, ko pārbauda jodometriski.
220.5. 3-merkaptopropān-1,2-diols (1-tioglicerīns), tīrība vismaz 98 %, ko pārbauda jodometriski.
220.6. Lietošanai gatavas 0,25 mm biezas silikagela plānslāņa plates.
220.7. Alumīnija oksīda, Merck F 254 E vai līdzvērtīgas plānslāņa plates.
220.8. Koncentrēta, ![]() = 1,19 g/ml,
sālsskābe.
= 1,19 g/ml,
sālsskābe.
220.9. Etilacetāts.
220.10. Hloroforms.
220.11. Diizopropilēteris.
220.12. Tetrahlorogleklis.
220.13. Ledus etiķskābe.
220.14. Kālija jodīds, 1 % (m/v) šķīdums ūdenī.
220.15. Platīna tetrahlorīds, 0,1 % (m/v) šķīdums ūdenī.
220.16. Eluenti:
220.16.1. Etilacetāts, hloroforms, diizopropilēteris, etiķskābe (20:20:10:10 tilp.v.).
220.16.2. Hloroforms, etiķskābe (90:20 tilp.v.).
220.17. Attīstītāji:
220.17.1. Tieši pirms lietošanas sajauc vienādus daudzumus šķīduma (atbilstoši šā pielikuma 220.14.apakšpunktam) un šķīduma (atbilstoši šā pielikuma 220.15.apakšpunktam).
220.17.2. Broma šķīdums, 5 % (m/v). Izšķīdina 5 g broma 100 ml tetrahloroglekļa.
220.17.3. Fluoresceīna šķīdums, 0,1 % (m/v). Izšķīdina 100 mg fluoresceīna 100 ml etanola.
220.17.4. Heksaamonija heptamolibdāts, 10 % (m/v) šķīdums ūdenī.
220.18. Standartšķīdumi:
220.18.1. Merkaptoetiķskābe (atbilstoši šā pielikuma 220.1.apakšpunktam), 0,4 % (m/v) šķīdums ūdenī.
220.18.2. 2,2'-ditiodi(etiķ)skābe (atbilstoši šā pielikuma 220.2.apakšpunktam), 0,4 % (m/v) šķīdums ūdenī.
220.18.3. 2-merkaptopropionskābe (atbilstoši šā pielikuma 220.3.apakšpunktam), 0,4 % (m/v) šķīdums ūdenī.
220.18.4. 3-merkaptopropionskābe (atbilstoši šā pielikuma 220.4.apakšpunktam), 0,4 % (m/v) šķīdums ūdenī.
220.18.5. 3-merkaptopropān-1,2-diols (atbilstoši šā pielikuma 220.5.apakšpunktam), 0,4 % (m/v) šķīdums ūdenī.
221. Merkaptoetiķskābes pierādīšanai plānslāņa hromatogrāfijā izmanto plānslāņa hromatogrāfijas standartaprīkojumu.
222. Merkaptoetiķskābes pierādīšanu plānslāņa hromatogrāfijā veic šādā kārtībā:
222.1. Paraugu apstrāde: skābina līdz pH 1 ar dažiem pilieniem sālsskābes (atbilstoši šā pielikuma 220.8.apakšpunktam) un pēc vajadzības filtrē. Dažos gadījumos var būt ieteicams paraugu atšķaidīt. Tādā gadījumā to pirms atšķaidīšanas skābina ar sālsskābi.
222.2. Eluēšana: uznes uz plates 1 μl parauga šķīduma (atbilstoši šā pielikuma 222.1.apakšpunktam) un vienu litru katra no pieciem standartšķīdumiem (220.18.apakšpunkts). Rūpīgi nožāvē vieglā slāpekļa plūsmā un eluē plati ar šķīdinātājiem (atbilstoši šā pielikuma 220.16.1. vai 220.16.2.apakšpunktam). Nožāvē plati iespējami ātri, lai mazinātu tiolu oksidāciju.
222.3. Attīstīšana: apsmidzina plati ar vienu no trijiem reaģentiem (atbilstoši šā pielikuma 220.17.1., 220.17.3. vai 220.17.4.apakšpunktam). Ja plati apsmidzina ar reaģentu (atbilstoši šā pielikuma 220.17.3.apakšpunktam), tad pēc tam to apstrādā ar broma tvaiku (tai skaitā kamerā, kur ir neliela vārglāze ar reaģentu (atbilstoši šā pielikuma 220.17.2.apakšpunktam)), līdz plankumi kļūst redzami. Attīstīšana ar uzsmidzināmo reaģentu (atbilstoši šā pielikuma 220.17.4.apakšpunktam) ir apmierinoša tikai tad, ja plānslāņa žāvēšanas laiks nepārsniedz 30 minūtes.
222.4. Interpretācija: salīdzina Rf vērtības un standartšķīdumu krāsu ar standartiem. Tabulā norādītās vidējās Rf vērtības izsaka tikai aptuvenu salīdzinājumu. Tās ir atkarīgas no plānslāņa aktivācijas stāvokļa hromatogrāfijas laikā un hromatogrāfijas kameras temperatūras.
Uz silikagela slāņa iegūto Rf vērtību piemēri
|
Eluenti |
||
|
atbilstoši šā pielikuma 220.16.1.apakšpunktam |
atbilstoši šā pielikuma 220.16.2.apakšpunktam |
|
|
Merkaptoetiķskābe |
0,25 |
0,80 |
|
2-merkaptopropionskābe |
0,40 |
0,95 |
|
2,2’-ditiodi(etiķ)skābe |
0,00 |
0,35 |
|
3-merkaptopropionskābe |
0,45 |
0,95 |
|
3-merkaptopropān-1,2-diols |
0,45 |
0,35 |
18.3. Merkaptoetiķskābes noteikšana. Jodometrija
223. Lai novērstu oksidēšanos, merkaptoetiķskābi nosaka nelietotam kosmētikas līdzeklim, kas ņemts no tikko atvērta trauka. Noteikšanu vienmēr sāk ar jodometriju.
224. Merkaptoetiķskābi nosaka, oksidējot –SH grupu ar jodu skābā vidē atbilstoši vienādojumam:
2 HOOC CH2SH + I2 (HOOC-CH2-S)2 + 2 I– + 2 H+
225. Mekaptoetiķskābes noteikšanai jodometrijā izmantojams jods, 0,05 M standartšķīdums.
226. Mekaptoetiķskābes noteikšanai jodometrijā izmantojams laboratorijas standartaprīkojums.
227. Mekaptoetiķskābes noteikšanu jodometrijā veic šādā kārtībā: precīzi iesver 0,5–1 g parauga 150 ml koniskajā kolbā ar aizbāzni, kur ir 50 ml destilēta ūdens. Pievieno 5 ml sālsskābes (šķīduma pH ap 0) un titrē ar joda šķīdumu (atbilstoši šā pielikuma 225.punktam), līdz parādās dzeltens krāsojums. Pēc izvēles lieto indikatoru (tai skaitā cietes šķīdumu vai tetrahloroglekli).
228. Merkaptoetiķskābes saturu aprēķina pēc šādas formulas:
% (m/m) = ![]() , kur
, kur
m – analizējamā parauga masa (gramos);
n – izmantotā joda šķīduma tilpums.
Ja merkaptoetiķskābes aprēķina rezultāts ir 0,1 % vai vairāk zem atļautās maksimālās koncentrācijas, nav vajadzības turpināt noteikšanu. Ja rezultāts ir vienāds ar vai augstāks par atļauto maksimālo koncentrāciju un pierādīšanā ir konstatēti vairāki reducējošie aģenti, ir nepieciešama noteikšana, veicot gāzu hromatogrāfiju.
18.4. Merkaptoetiķskābes noteikšana. Gāzu hromatogrāfija
229. Merkaptoetiķskābi atdala no palīgvielas, nogulsnējot ar kadmija diacetāta šķīdumu. Pēc metilēšanas ar diazometānu, ko gatavo in situ vai iepriekš dietilētera šķīdumā, merkaptoetiķskābes metilatvasinājumu mēra gāzu/šķidruma hromatogrāfijā, par iekšējo standartu izmantojot metiloktanoātu.
230. Merkaptoetiķskābes noteikšanai gāzu hromatogrāfijā izmanto šādus reaģentus1:
230.1. Merkaptoetiķskābe, 98 %.
230.2. Sālsskābe, ![]() = 1,19 g/ml.
= 1,19 g/ml.
230.3. Metanols.
230.4. Kadmija diacetāta dihidrāts, 10 % (m/v) šķīdums ūdenī.
230.5. Metiloktanoāts, 2 % (m/v) šķīdums metanolā.
230.6. Acetāta buferšķīdums (pH 5):
230.6.1. Nātrija acetāta trihidrāts, 77 g.
230.6.2. Etiķskābe (ledus), 27,5 g.
230.6.3. Demineralizēts ūdens, ar ko uzpilda līdz vienam litram.
230.7. Sālsskābe, svaigi sagatavots 3 M šķīdums metanolā.
230.8. 1-metil-3-nitro-1-nitrozoguanidīns.
230.9. Nātrija hidroksīds, 5 M šķīdums.
230.10. Jods, 0,05 M standartšķīdums.
230.11. Dietilēteris.
230.12. Diazometāna šķīdums, kas gatavots no N-metil-N-nitrozotoluol-4-sulfonamīda (Fieser, Reagents for Organic Synthesis (Wiley), 1967).Iegūtais šķīdums satur apmēram 1,5 g diazometāna 100 mililitros dietilētera. Tā kā diazometāns ir toksiska un ļoti nestabila gāze, visi eksperimenti jāizdara stiprā velkmē un jāizvairās lietot slīpēta stikla aprīkojumu (šim nolūkam ir īpaši komplekti).
231. Merkaptoetiķskābes noteikšanai gāzu hromatogrāfijā izmanto šādu aprīkojumu.
231.1. Laboratorijas standartaprīkojums.
231.2. Iekārta diazometāna gatavošanai in situ metilēšanai (Fales, H.M., Jaouni, T.M. and Babashak, J.F., Analyt. Chem. 1973, 45, 2302).
231.3. Iekārta iepriekšējai diazometāna gatavošanai (Fieser).
232. Parauga gatavošanu merkaptoetiķskābes noteikšanai gāzu hromatogrāfijā veic šādā kārtībā:
232.1. Precīzi iesver 50 ml centrifūgas mēģenē pietiekami daudz parauga, lai iegūtu iepriekš pieņemtu 50–70 mg daudzumu merkaptoetiķskābes.
232.2. Paskābina ar dažiem pilieniem sālsskābes (atbilstoši šā pielikuma 230.2.apakšpunktam), lai iegūtu aptuvenu pH 3.
232.3. Pievieno 5 ml demineralizēta ūdens un 10 ml acetāta buferšķīduma (atbilstoši šā pielikuma 230.6.apakšpunktam) un ar pH papīru pārbauda, vai pH vērtība ir aptuveni 5.
232.4. Pievieno 5 ml kadmija diacetāta šķīduma (atbilstoši šā pielikuma 230.4.apakšpunktam). Pagaida 10 minūtes.
232.5. Centrifugē vismaz 15 minūtes ar 4000 apgr./min.
232.6. Atdala centrifugātu, kurā var būt nešķīstoši tauki (ja paraugs ņemts no krēma). Šos taukus nevar sajaukt ar tioliem, kas sakrājas kompaktā masā mēģenes apakšā.
232.7. Pārbauda, vai neveidojas nogulsnes, ja centrifugātam pievieno dažus pilienus kadmija diacetāta šķīduma (atbilstoši šā pielikuma 230.4.apakšpunktam).
232.8. Ja iepriekšējās pierādīšanās nav konstatēti reducējoši aģenti, izņemot tiolus, jodometriski pārbauda, vai centrifugātā esošais tiols nepārsniedz 6–8 % sākotnējā daudzuma.
232.9. Ievada 10 ml metanola centrifūgas mēģenē, kur ir nogulsnes, un viegli disperģē nogulsnes ar stikla spieķīti. Centrifugē vēlreiz vismaz 15 minūtes ar 4000 apgr./min.
232.10. Mazgā nogulsnes otrreiz, ievērojot iepriekš minēto procedūru. Tajā pašā centrifūgas mēģenē pievieno 2 ml metiloktanoāta šķīduma (atbilstoši šā pielikuma 230.5.apakšpunktam0) un 5 ml sālsskābes šķīduma metanolā (atbilstoši šā pielikuma 230.7.apakšpunktam).
232.11. Pilnīgi izšķīdina tiolus (var palikt nedaudz nešķīstošas palīgvielas). Šis ir šķīdums S. Ar šī šķīduma alikvoto daļu jodometriski pārbauda, vai tiolu saturs ir vismaz 90 % no tā, kas iegūts saskaņā ar šā pielikuma 18.3.apakšnodaļu.
233. Metilē, gatavojot in situ (atbilstoši šā pielikuma 231.2.apakšpunktam) vai ar iepriekš gatavotu diazometāna šķīdumu (atbilstoši šā pielikuma 231.3.apakšpunktam):
233.1. Metilēšana in situ. Metilēšanas iekārtā, kur ir 1 ml dietilētera, ievada 50 μl S šķīduma un metilē pēc metodes (atbilstoši šā pielikuma 231.2.apakšpunktam) ar 300 mg 1-metil-3-nitro-1-nitrozoguanidīna. Pēc 15 minūtēm (ētera šķīdumam vajadzētu būt dzeltenam, kas liecina par diazometāna pārākumu) parauga šķīdumu pārnes uz 2 ml pudeli, kurai ir blīvs aizbāznis. Uz nakti liek ledusskapī. Metilē divus paraugus vienlaicīgi.
233.2. Metilēšana ar iepriekš gatavoto diazometāna šķīdumu. Ievada 5 ml kolbā ar aizbāzni 1 ml diazometāna šķīduma (atbilstoši šā pielikuma 230.12.apakšpunktam), pēc tam 50 μl S šķīduma. Uz nakti liek ledusskapī.
234. Standartu gatavo šādi: gatavo merkaptoetiķskābes standartšķīdumu, kas satur apmēram 60 mg tīras merkaptoetiķskābes 2 mililitros. Šis ir šķīdums E. Nogulsnē, pārbauda un metilē saskaņā ar šā pielikuma 231., 232. un 233.punktu.
235. Gāzu hromatogrāfijas apstākļi merkaptoetiķskābes noteikšanai:
235.1. Kolonna:
235.1.1. Tips: nerūsējošs tērauds.
235.1.2. Garums: 2 m.
235.1.3. Diametrs: 3 mm.
235.2. Pildījums: 20 % didecilftalāts/hromosorbs, WAW ar daļiņu lielumu 80–100.
235.3. Piemērots ir liesmas jonizācijas detektors ar elektrometra jutības uzstādījumu 8 × 10-10 A.
235.4. Gāze:
235.4.1. Nesējgāze: slāpeklis.
235.4.2. Spiediens: 2,2 bar.
235.4.3. Plūsma: 35 ml/min.
235.4.4. Palīggāze: ūdeņradis.
235.4.5. Spiediens: 1,8 bar.
235.4.6. Plūsma: 15 ml/min.
235.4.7. Pievade detektoram: pēc iekārtas izgatavotāju norādījumiem.
235.5. Temperatūra:
235.5.1. Inžektors: 200 °C.
235.5.2. Detektors: 200 °C.
235.5.3. Kolonna: 90 °C.
235.6. Pašrakstītāja ātrums: 5 mm/min.
235.7. Ievadītais daudzums: 3 μl. Ievada piecas reizes.
235.8. Hromatogrāfijas apstākļi ir orientējoši. Tie dod iespēju nodrošināt izšķirtspēju R, kas ir vienāda ar 1,5 vai labāka:
![]() , kur
, kur
r1 un r2 – izdalīšanas laiki (minūtēs);
W1 un W2 – pīķu platumi pusaugstumā (milimetros);
d' – pašrakstītāja ātrums (milimetros minūtē).
Ieteicams hromatogrāfiju pārtraukt, noregulējot temperatūru no 90 uz 150 °C pa 10 °C minūtē, lai likvidētu vielas, kas var traucēt turpmākos mērījumus.
236. Aprēķinus veic šādā kārtībā:
236.1. Merkaptoetiķskābes proporcionalitātes koeficients. To aprēķina attiecībā pret metiloktanoātu, pamatojoties uz standartmaisījumu. Ja merkaptoetiķskābi apzīmē ar t, tad:
kt – tās signāla attiecība pret masas vienību;
m't – tās masa (miligramos) maisījumā;
S't – tās pīķa laukums.
Ja metiloktanoātu apzīmē ar c, tad:
m'c – tā masa (miligramos) maisījumā;
S'c – tā pīķa laukums, un:
Šis koeficients atšķiras atkarībā no iekārtas.
236.2. Merkaptoetiķskābes koncentrācija paraugā. Ja merkaptoetiķskābi apzīmē ar t, tad:
kt – tās signāla attiecība pret masas vienību;
St – tās pīķa laukums.
Ja metiloktanoātu apzīmē ar c, tad:
mc – tā masa (miligramos) maisījumā;
Sc – tā pīķa laukums;
M – sākotnējā analizējamā parauga masa (miligramos), un:
merkaptoetiķskābes % (m/m) paraugā ir: ![]()
237. Ja merkaptoetiķskābes saturs ir 8 % (m/m), viena parauga divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,8 % (m/m) vērtību4.
19. Heksahlorofēna pierādīšana un noteikšana
19.1. Heksahlorofēna pierādīšana
238. Heksahlorfēna pierādīšanas metode ir piemērota visiem kosmētikas līdzekļiem.
239. Heksahlorofēnu paraugā ekstrahē ar etilacetātu un pierāda plānslāņa hromatogrāfijā.
240. Heksahlorofēna pierādīšanas metodei izmanto šādus reaģentus1:
240.1. Sērskābe, 4 M šķīdums.
240.2. Celīts AW.
240.3. Etilacetāts.
240.4. Eluents: benzols, kas satur 1 % (v/v) ledus etiķskābes.
240.5. Attīstītājs I ir rodamīna B šķīdums, ko pagatavo šādi: izšķīdina 100 mg rodamīna B maisījumā, kas satur 150 ml dietilētera, 70 ml absolūtā etanola un 16 ml ūdens.
240.6. Attīstītājs II ir 2,6-dibrom-4-hloriminocikloheksa-2,5-dienona šķīdums, ko pagatavo šādi: izšķīdina 400 mg 2,6-dibrom-4-hloriminocikloheksa-2,5-dienona 100 ml metanola (katru dienu gatavo svaigu).
Nātrija karbonāta šķīdums: izšķīdina 10 g nātrija karbonāta 100 ml demineralizēta ūdens.
240.7. Standartšķīdums: Heksahlorofēns, 0,05 % (m/v) šķīduma etilacetātā.
241. Heksahlorofēna pierādīšanas metodei izmanto šādu aprīkojumu:
241.1. Silikagela 254 plānslāņa hromatogrāfijas plates, 200 × 200 mm (vai līdzvērtīgas).
241.2. Plānslāņa hromatogrāfijas standartaprīkojums.
241.3. Vanna, kurā ar termostatu uzstāda 26 °C, hromatogrāfijas kamerai.
242. Analizējamā parauga sagatavošana heksahlorofēna pierādīšanai:
242.1. Labi sajauc 1 g homogenizēta parauga ar 1 g celīta AW un 1 ml sērskābes (atbilstoši šā pielikuma 240.1.apakšpunktam).
242.2. Žāvē 100 °C divas stundas.
242.3. Atdzesē un izžuvušo atlikumu saberž pulverī.
242.4. Divas reizes ekstrahē, katru reizi lietojot 10 ml etilacetāta, pēc katras ekstrakcijas centrifugē un apvieno etilacetāta slāņus.
242.5. Tvaicē 60 °C.
242.6. Izšķīdina atlikumu 2 ml etilacetāta.
243. Heksahlorofēna pierādīšanu veic šādā kārtībā:
243.1. Uznes 2 μl analizējamā parauga šķīduma (atbilstoši šā pielikuma 242.6.apakšpunktam) un 2 μl standartšķīduma (atbilstoši šā pielikuma 240.7.apakšpunktam) uz plānslāņa hromatogrāfijas plates.
243.2. Piesātina kameru (atbilstoši šā pielikuma 241.3.apakšpunktam) ar eluentu (atbilstoši šā pielikuma 240.4.apakšpunktam).
243.3. Liek plānslāņa hromatogrāfijas plati kamerā un eluē līdz 150 mm.
243.4. Izņem plānslāņa hromatogrāfijasplati un žāvē ventilācijas tipa žāvējamā skapī apmēram 105 °C temperatūrā.
243.5. Heksahlorofēna plankumus uz plānslāņa plates attīsta saskaņā ar šā pielikuma 243.5.1. vai 243.5.2.apakšpunktu:
243.5.1. Plati vienmērīgi apsmidzina ar attīstītāju I (atbilstoši šā pielikuma 240.5.apakšpunktam). Pēc 30 minūtēm plati apskata ultravioletā gaismā 254 nm.
243.5.2. Plati vienmērīgi apsmidzina ar attīstītāja II (atbilstoši šā pielikuma 240.6.apakšpunktam) 2,6-dibrom-4-hloriminocikloheksa-2,5-dienona šķīdumu. Pēc tam plati apsmidzina ar nātrija karbonāta šķīdumu (atbilstoši šā pielikuma 240.6.apakšpunktam), žāvē 10 minūtes istabas temperatūrā un apskata dienasgaismā.
244. Interpretācija:
244.1. Attīstītājs I (atbilstoši šā pielikuma 240.5.apakšpunktam): heksahlorofēns parādās zilgana plankuma veidā uz dzelteni oranža fluorescējoša fona un tā Rf vērtība ir aptuveni 0,5.
244.2. Attīstītājs II (atbilstoši šā pielikuma 240.6.apakšpunktam): heksahlorofēns parādās kā debeszilas līdz tirkīzzilas krāsas plankums uz balta fona, un tā Rf vērtība ir aptuveni 0,5.
19.2. Heksahlorofēna noteikšana
245. Heksahlorofēna noteikšanas metode piemērojama visiem kosmētikas līdzekļiem.
246. Heksahlorofēna saturu paraugā izsaka heksahlorofēna masas procentos.
247. Heksahlorofēnu pēc konversijas metilatvasinājumā nosaka, veicot gāzu hromatogrāfiju ar elektronu satveres detektoru.
248. Heksahlorofēna noteikšanas metodei izmanto šādus reaģentus1:
248.1. Etilacetāts.
248.2. N-metil-N-nitrozo-p-toluolsulfonamīds (diazalds).
248.3. Dietilēteris.
248.4. Metanols.
248.5. 2-(2-etoksietoksi)etanols (karbitols).
248.6. Skudrskābe.
248.7. Kālija hidroksīds, 50 % (m/m) ūdens šķīdums (katru dienu gatavo svaigu).
248.8. Heksāns spektroskopijai.
248.9. Bromhlorofēns (standarts Nr.1).
248.10. 4,4',6,6'-tetrahlor-2,2'-tiodifenols (standarts Nr.2).
248.11. 2,4,4'-trihlor-2-hidroksidifenilēteris (standarts Nr.3).
248.12. Acetons.
248.13. 4 M sērskābe.
248.14. Celīts AW.
248.15. Skudrskābe/etilacetāts, 10 % (v/v) šķīdums.
248.16. Heksahlorofēns.
249. Heksahlorofēna noteikšanas metodei izmanto šādu aprīkojumu:
249.1. Laboratorijas standarta stikla trauki.
249.2. Mikroiekārta diazometāna gatavošanai (Analyt.Chem., 1973, 45, 2302-2).
249.3. Gāzes hromatogrāfs, kas aprīkots ar 63 Ni avota elektronu satveres detektoru.
250. Heksahlorofēna noteikšanu veic šādā kārtībā:
250.1. Standartšķīduma gatavošanai standartu izvēlas tādu, kas neiedarbojas ne uz vienu palīgvielu analizējamā kosmētikas līdzeklī. Parasti piemērotākais ir standarts Nr.1 (atbilstoši šā pielikuma 248.9.apakšpunktam):
250.1.1. Precīzi iesver aptuveni 50 mg standarta Nr.1, 2 vai 3 (atbilstoši šā pielikuma 248.9., 248.10. vai 248.11.apakšpunktam) un 50 mg heksahlorofēna 100 ml mērkolbā. Uzpilda līdz zīmei ar etilacetātu (turpmāk -šķīdums A). Atšķaida 10 ml šķīduma A līdz 100 ml ar etilacetātu (turpmāk - šķīdums B).
250.1.2. Precīzi iesver aptuveni 50 mg standarta Nr.1, 2 vai 3 (atbilstoši šā pielikuma 248.9., 248.10. vai 248.11.apakšpunktam) 100 ml mērkolbā. Uzpilda līdz zīmei ar etilacetātu (turpmāk - šķīdums C).
250.2. Parauga gatavošana: precīzi nosver 1 g homogenizēta parauga un rūpīgi sajauc ar 1 ml sērskābes (atbilstoši šā pielikuma 248.13.apakšpunktam), 15 ml acetona un 8 g celīta AW. Žāvē maisījumu gaisā 30 minūtes tvaika vannā, pēc tam pusotru stundu žāvē ventilācijas tipa žāvēšanas skapī. Atdzesē, atlikumu saberž pulverī un pārnes uz stikla kolonnu. Eluē ar etilacetātu un savāc 100 ml. Pievieno 2 ml iekšējā standarta šķīduma (šķīdums C) (atbilstoši šā pielikuma 250.1.2.apakšpunktam). Tā kā heksahlorofēns varētu būt dažādu veidu kosmētikas līdzekļos, ir svarīgi pārbaudīt heksahlorofēna rekuperāciju no parauga šajā procedūrā pirms rezultātu reģistrēšanas. Ja rekuperācija ir zema, tad, ieinteresētajām pusēm vienojoties, izmaina procedūru (piemēram, lietojot citu šķīdinātāju (benzolu etilacetāta vietā) u.tml.).
250.3. Parauga metilēšana: dzesē visus reaģentus un iekārtu līdz 0–4 °C divas stundas. Diazometāna iekārtas ārējā nodalījumā liek 1,2 ml šķīduma, ko gatavo saskaņā ar šā pielikuma 250.2.apakšpunktu un 0,1 ml metanola. Liek aptuveni 200 mg diazalda centrālajā rezervuārā, pievieno 1 ml karbitola, 1 ml dietilētera un izšķīdina. Samontē iekārtu, līdz pusei iegremdē to vannā 0 °C un centrālajā rezervuārā ar šļirci ievada apmēram 1 ml atdzesēta kālija hidroksīda šķīduma (atbilstoši šā pielikuma 248.7.apakšpunktam). Nodrošina to, ka dzeltenais krāsojums, kas rodas, veidojoties diazometānam, saglabājas. Ja dzeltenais krāsojums nesaglabājas, metilēšanu atkārto vēl ar 200 mg diazalda. Ja dzeltenais krāsojums saglabājas, tas liecina par diazometāna pārākumu, kas nepieciešams, lai nodrošinātu pilnīgu parauga metilēšanu. Iekārtu izņem no vannas pēc 15 minūtēm un atstāj noslēgtu apkārtējās vides temperatūrā uz 12 stundām. Atver iekārtu, pievieno dažus pilienus 10 % (v/v) skudrskābes šķīduma etilacetātā (atbilstoši šā pielikuma 248.15.apakšpunktam), lai izreaģē diazometāna pārākums, un pārnes organisko šķīdumu uz 25 ml mērkolbu. Uzpilda līdz zīmei ar heksānu. Ievada 1,5 μl sagatavotā šķīduma hromatogrāfā.
250.4. Standarta metilēšana: dzesē visus reaģentus un iekārtu līdz 0–4 °C divas stundas. Diazometāna iekārtas ārējā nodalījumā ievada: 0,2 ml šķīduma B (atbilstoši šā pielikuma 250.1.1.apakšpunktam), 1 ml etilacetāta un 0,1 ml metanola. Turpina metilēt saskaņā ar šā pielikuma 250.3.apakšpunktu. Ievada 1,5 μl iegūtā šķīduma hromatogrāfā.
251. Gāzu hromatogrāfiju heksahlorofēna noteikšanai veic šādā kārtībā: kolonnai jānodrošina izšķirtspēja R, kas ir 1,5 vai labāka:
![]() , kur
, kur
r1 un r2 – izdalīšanas laiki (minūtēs);
W1 un W2 – pīķu platumi pusaugstumā (milimetros);
d' – pašrakstītāja ātrums (milimetros minūtē).
251.1. Par piemērotiem uzskata šādus gāzu hromatogrāfijas apstākļus:
251.1.1. Kolonna: nerūsējošs tērauds:
251.1.1.1. Garums: 1,7 m, diametrs: 3 mm.
251.1.1.2. Nesējs: hromosorbs: WAW.
251.1.2. Sieta analīze: daļiņu lielums 80–100.
251.1.3. Nekustīgā fāze: 10 % OV 17.
251.1.4. Temperatūra:
251.1.4.1. Kolonna: 280 °C.
251.1.4.2. Inžektors: 280 °C.
251.1.4.3. Detektors: 280 °C.
251.1.5. Nesējgāze: slāpeklis bez skābekļa.
251.1.6. Spiediens 2,3 bar.
251.1.7. Plūsma: 30 ml/min.
252. Aprēķinus veic šādā kārtībā:
252.1. Heksahlorofēna proporcionalitātes koeficients. To atkarībā no izvēlētā standarta aprēķina attiecībā pret standarta maisījumu. Ja:
h – heksahlorofēns;
kh – tā proporcionalitātes koeficients;
m'h – tā masa (gramos) maisījumā;
A'h – tā pīķa laukums;
s – izvēlētais standarts;
m's – tā masa (gramos) maisījumā;
A's – tā pīķa laukums, tad
252.2. Heksahlorofēna daudzums paraugā. Ja:
h – heksahlorofēns;
kh – tā proporcionalitātes koeficients;
Ah – tās pīķa laukums;
s – izvēlētais standarts;
ms – tā masa (gramos) maisījumā;
As – tā pīķa laukums;
M – ņemtā parauga masa (gramos), tad:
% (m/m) heksahlorofēna paraugā ir: ![]()
253. No viena parauga ar 0,1 % (m/m) heksahlorofēna saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,005 % (m/m) vērtību4.
20. Nātrija tozilhloramīda (INN) (hloramīna-T) kvantitatīvā noteikšana
254. Ar hloramīna-T kvantitatīvās noteikšanas metodi kvantitatīvi nosaka nātrija tozilhloramīdu (hloramīnu-T) kosmētikas līdzekļos, veicot plānslāņa hromatogrāfiju.
255. Hloramīna-T saturu paraugā izsaka masas procentos (m/m).
256. Hloramīnu-T pilnīgi hidrolizē līdz 4-toluolsulfonamīdam, vārot kopā ar sālsskābi. Radušos 4-toluolsulfonamīda daudzumu fotodensitometriski nosaka plānslāņa hromatogrāfijā.
257. Hloramīna-T kvantitatīvās noteikšanas metodei izmanto šādus reaģentus1:
257.1. Nātrija tozilhloramīds (hloramīns-T).
257.2. 4-toluolsulfonamīda standartšķīdums: 50 mg 4-toluolsulfonamīda 100 ml etanola (atbilstoši šā pielikuma 257.5.apakšpunktam).
257.3. Sālsskābe, 37 % (m/m), ![]() = 1,18 g/ml.
= 1,18 g/ml.
257.4. Dietilēteris.
257.5. Etanols, 96 % (v/v).
257.6. Attīstīšanas šķīdinātājs:
257.6.1. 1-butanols/etanols (atbilstoši šā pielikuma 257.5.apakšpunktam)/ūdens 40:4:9 (v/v/v).
257.6.2. Hloroforms/acetons 6:4:4 (v/v).
257.7. Lietošanai gatavas plānslāņa hromatogrāfijas plates, silikagels 60, bez fluorescējoša indikatora.
257.8. Kālija permanganāts.
257.9. Sālsskābe, 15 % (m/m).
257.10. Izsmidzināmais reaģents: 2-toluidīns, 1 % (m/v) šķīdums etanolā (atbilstoši šā pielikuma 257.5.apakšpunktam).
258. Hloramīna-T kvantitatīvās noteikšanas metodei izmanto šādu aprīkojumu:
258.1. Laboratorijas standartaprīkojums.
258.2. Standarta plānslāņa hromatogrāfijas iekārta.
258.3. Fotodensitometrs.
259. Hloramīna-T kvantitatīvo noteikšanu veic šādā kārtībā:
259.1. Hidrolīze. Precīzi iesver 50 ml apaļkolbā aptuveni 1 g parauga (m). Pievieno 5 ml ūdens un 5 ml sālsskābes (atbilstoši šā pielikuma 257.3.apakšpunktam) un vāra vienu stundu, izmantojot atteces dzesinātāju. Karsto suspensiju ar ūdeni tūlīt pārnes uz 50 ml mērkolbu. Ļauj atdzist un uzpilda līdz zīmei ar ūdeni. Centrifugē piecas minūtes vismaz ar 3000 apgriezieniem minūtē un izfiltrē centrifugātu.
259.2. Ekstrakcija:
259.2.1. Ņem 30 ml filtrāta un ekstrahē trīs reizes ar 15 ml dietilētera. Pēc vajadzības žāvē ētera fāzes un savāc tās 50 ml mērkolbā. Uzpilda ar dietilēteri.
259.2.2. Ņem 25 ml žāvētā ētera ekstrakta un ietvaicē līdz sausam stāvoklim slāpekļa atmosfērā. Vēlreiz izšķīdina atlikumu 1 ml etanola.
259.3. Plānslāņa hromatogrāfija:
259.3.1. Uznes 20 μl etanola atlikuma (atbilstoši šā pielikuma 259.2.apakšpunktam) uz plānslāņa hromatogrāfijas plates (atbilstoši šā pielikuma 257.7.apakšpunktam). Vienlaicīgi tāpat klāj 8, 12, 16 un 20 μl 4-toluolsulfonamīda standartšķīduma (atbilstoši šā pielikuma 257.2.apakšpunktam).
259.3.2. Ļauj attīstīties apmēram 150 mm attīstīšanas šķīdinātājā (atbilstoši šā pielikuma 257.6.1. vai 257.6.2.apakšpunktam).
259.3.3. Kad attīstīšanas šķīdinātājs ir pilnīgi ietvaicēts, liek plati uz divām vai trijām minūtēm hlora tvaika atmosfērā, ko rada, slēgtā traukā uzlejot apmēram 100 ml sālsskābes (atbilstoši šā pielikuma 257.9.apakšpunktam) uz apmēram 2 g kālija permanganāta. Pārākuma hloru atdala, piecas minūtes karsējot plati līdz 100 °C. Pēc tam plati apsmidzina ar reaģentu (atbilstoši šā pielikuma 257.10.apakšpunktam).
259.4. Mērīšana: apmēram pēc vienas stundas ar fotodensitometru 525 nm izmēra violetos plankumus.
259.5. Kalibrēšanas līkņu konstruēšana: atzīmē pīķu maksimālo augstumu vērtības atbilstoši četriem 4-toluolsulfonamīda plankumiem pret attiecīgajiem 4-toluolsulfonamīda daudzumiem (piemēram, 4, 6, 8, 10 μg 4-toluolsulfonamīda uz vienu plankumu).
260. Metodi var kontrolēt ar 0,1 vai 0,2 % (m/v) hloramīna-T šķīdumu, ko apstrādā tāpat kā paraugu (atbilstoši šā pielikuma 259.punktam).
261. Hloramīna-T saturu, kas izteikts masas procentos, paraugā aprēķina šādi:
% (m/m) tozilhloramīda = ![]() , kur
, kur
1,33 – 4-toluolsulfonamīda-hloramīna-T konversijas koeficients;
a – 4-toluolsulfonamīda daudzums (μg) paraugā saskaņā ar kalibrēšanas līknēm;
m – ņemtā parauga masa (gramos).
262. No viena parauga ar 0,2 % (m/m) hloramīna-T saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,03 % (m/m) vērtību4.
21. Kopējā fluora noteikšana zobu pastās
263. Ar kopējā fluora noteikšanas zobu pastās metodi nosaka kopējā fluora daudzumu zobu pastās, ja fluora līmenis nepārsniedz 0,25 %.
264. Fluora saturu paraugā izsaka masas procentos.
265. Kopējā fluora noteikšanu veic gāzes hromatogrāfijā. Fluoru, kas ir fluoru saturošajos savienojumos, pārvērš trietilfluorsilānā (TEFS)tiešā reakcijā ar hlortrietilsilānu (TECS) skābes šķīdumā un vienlaicīgi ekstrahē ar ksilolu saturošu cikloheksānu kā iekšējo standartu.
266. Kopējā fluora noteikšanas zobu pastās metodei izmanto šādus reaģentus1:
266.1. Nātrija fluorīds, kas 120 °C izžāvēts līdz nemainīgai masai.
266.2. Ūdens, divreiz destilēts vai līdzvērtīgas kvalitātes.
266.3. Sālsskābe, ![]() = 1,19 g/ml.
= 1,19 g/ml.
266.4. Cikloheksāns (CH).
266.5. Ksilols, kam hromatogrammā nav pīķu pirms šķīdinātāja pīķa, ja hromatografē tādos pašos apstākļos kā paraugu (atbilstoši šā pielikuma 268.1.apakšpunkts). Ja nepieciešams, attīra destilējot (267.8.apakšpunktam).
266.6. Hlortrietilsilāns (TECS Merck vai līdzvērtīgs).
266.7. Fluora standartšķīdumi:
266.7.1. Standartšķīdums. 0,250 mg F-/ml. Precīzi nosver 138,1 mg nātrija fluorīda (atbilstoši šā pielikuma 266.1.apakšpunktam) un izšķīdina ūdenī (atbilstoši šā pielikuma 266.2.apakšpunktam). Kvantitatīvi pārnes šķīdumu uz 250 ml mērkolbu (atbilstoši šā pielikuma 267.5.apakšpunktam). Atšķaida ar ūdeni (atbilstoši šā pielikuma 266.2.apakšpunktam) līdz atzīmei un sajauc.
266.7.2. Atšķaidīts standartšķīdums, 0,050 mg F-/ml. Ar pipeti pārnes 20 ml standartšķīduma (atbilstoši šā pielikuma 266.7.1.apakšpunktam) uz 100 ml mērkolbu (atbilstoši šā pielikuma 267.5.apakšpunktam). Atšķaida ar ūdeni līdz atzīmei un sajauc.
266.8. Iekšējā standarta šķīdums: sajauc 1 ml cikloheksāna un 5 ml ksilola (atbilstoši šā pielikuma 266.5.apakšpunktam).
266.9. Hlortrietilsilāns/iekšējā standarta šķīdums: ar pipeti (atbilstoši šā pielikuma 267.7.apakšpunktam) pārnes 0,6 ml TECS un 0,12 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 266.8.apakšpunktam) uz 10 ml mērkolbu. Atšķaida ar ksilolu (atbilstoši šā pielikuma 266.5.apakšpunktam) līdz zīmei un sajauc. Katru dienu gatavo svaigu šķīdumu.
266.10. Perhlorskābe, 70 % (m/v).
266.11. Perhlorskābe, 20 % (m/v) ūdenī (atbilstoši šā pielikuma 266.2.apakšpunktam).
267. Kopējā fluora noteikšanas zobu pastās metodei izmanto šādu aprīkojumu:
267.1. Laboratorijas standartaprīkojums.
267.2. Gāzu hromatogrāfs, kas aprīkots ar liesmas jonizācijas detektoru.
267.3. Virpuļmaisītājs vai līdzvērtīgs.
267.4. Kratītājs, SMB1 tipa vai līdzvērtīgs.
267.5. Mērkolbas, 100 un 250 ml, kas izgatavotas no polipropilēna.
267.6. Centrifūgas mēģenes (stikla); 20 ml, kam ir ar teflonu pārklāti skrūvējami vāciņi, Sovirel 611-56 vai līdzvērtīgas. Iztīra mēģenes un skrūvējamos vāciņus, vairākas stundas turot perhlorskābē (atbilstoši šā pielikuma 266.11.apakšpunktam), pēc tam piecas reizes skalo ar ūdeni (atbilstoši šā pielikuma 266.2.apakšpunktam) un beidzot žāvē 100 °C.
267.7. Pipetes, ko var noregulēt 50–200 μl tilpuma pārnešanai, ar vienreizējas lietošanas plastmasas uzgaļiem.
267.8. Destilācijas iekārta, kam ir Šneidera kolonna ar trim lodītēm vai līdzvērtīga Vigrē kolonna.
268. Kopējā fluora noteikšanu veic šādā kārtībā:
268.1. Parauga analīze:
268.1.1. Ņem zobu pastas tūbiņu, kas iepriekš nav bijusi atvērta, atver un izņem visu saturu. Pārnes uz plastikas trauku, labi sajauc un glabā tādos apstākļos, kuros pasta neizmainās.
268.1.2. Precīzi iesver 150 mg (m) parauga centrifūgas mēģenē (atbilstoši šā pielikuma 267.6.apakšpunktam), pievieno 5 ml ūdens (atbilstoši šā pielikuma 266.2.apakšpunktam) un homogenizē virpuļmaisītājā.
268.1.3. Pievieno 1 ml ksilola.
268.1.4. Pa pilienam pievieno 5 ml sālsskābes (atbilstoši šā pielikuma 266.3.apakšpunktam) un homogenizē virpuļmaisītājā.
268.1.5. Ar pipeti centrifūgas mēģenē (atbilstoši šā pielikuma 267.6.apakšpunktam) pievieno 0,5 ml hlortrietilsilāna/iekšējā standarta šķīduma (atbilstoši šā pielikuma 266.9.apakšpunktam).
268.1.6. Noslēdz mēģeni ar skrūvējamo vāciņu (atbilstoši šā pielikuma 267.6.apakšpunktam) un rūpīgi jauc 45 minūtes ar kratītāju, kas noregulēts uz 150 gājieniem minūtē.
268.1.7. Centrifugē 10 minūtes tādā ātrumā, lai fāzes skaidri atdalās, atskrūvē mēģeni, izņem organisko slāni un ievada 3 μl organiskās fāzes gāzu hromatogrāfa kolonnā. Apmēram pēc 20 minūtēm visi komponenti ir eluēti.
268.1.8. Ievada vēlreiz, aprēķina vidējo pīķu laukumu attiecību (ATEFS/ACH) un no kalibrēšanas līknes (atbilstoši šā pielikuma 268.3.apakšpunktam) nolasa attiecīgo fluora daudzumu (miligramos (m1)).
268.1.9. Aprēķina kopējo fluora saturu paraugā (fluora masas procentos), kā norādīts šā pielikuma 269.apakšpunktā.
268.2. Hromatogrāfijas apstākļi:
268.2.1. Kolonna: nerūsējošs tērauds.
268.2.1.1. Garums: 1,8 m.
268.2.1.2. Diametrs: 3 mm.
268.2.1.3. Nesējs: Gaschrom Q ar daļiņu lielumu 80–100.
268.2.1.4. Nekustīgā fāze: silikoneļļa DC 200 vai līdzvērtīga, 20 %. Pa nakti kondicionē kolonnu 100 °C (nesējgāzes plūsma: 25 ml slāpekļa minūtē), to atkārto katru nakti. Pēc katras ceturtās vai piektās ievadīšanas kolonnu kondicionē atkārtoti, karsējot 30 minūtes 100 °C.
268.2.2. Temperatūra:
268.2.2.1. kolonna: 70 °C.
268.2.2.2. inžektors: 150 °C.
268.2.2.3. detektors: 250 °C.
268.2.3. Nesējgāzes plūsma: 35 ml slāpekļa minūtē.
268.3. Kalibrēšanas līkne:
268.3.1. Ar pipeti sešās centrifūgas mēģenēs (atbilstoši šā pielikuma 267.6.apakšpunktam) iepilina 0, 1, 2, 3, 4 un 5 ml atšķaidīta fluorīda standartšķīduma (atbilstoši šā pielikuma 266.7.2.apakšpunktam). Visas mēģenes uzpilda līdz 5 ml ar ūdeni (atbilstoši šā pielikuma 266.2.apakšpunktam).
268.3.2. Turpina darbību saskaņā ar šā pielikuma 268.1.3., 268.1.4., 268.1.5. un 268.1.6.apakšpunktu.
268.3.3. Ievada 3 μl organiskas fāzes gāzu hromatogrāfa kolonnā.
268.3.4. Ievada atkārtoti un aprēķina vidējo pīķu attiecību (ATEFS/ACH).
268.3.5. Konstruē kalibrēšanas līkni, salīdzinot fluora masu (miligramos) standartšķīdumos (atbilstoši šā pielikuma 268.3.1.apakšpunktam) un pīķu laukumu attiecību (ATEFS/ACH), ko mēra saskaņā ar šī pielikuma 268.3.4.apakšpunktu. Līknes punktus savieno ar atbilstīgāko taisno līniju, ko aprēķina pēc regresijas analīzes.
269. Kopējā fluora koncentrāciju paraugā (fluora masas procentos) (% (m/m) F) aprēķina pēc formulas:
% F = ![]() , kur
, kur
m – parauga daudzums (miligramos) (atbilstoši šā pielikuma 268.1.2.apakšpunktam);
m1 – F daudzums (miligramos), ko nolasa no kalibrēšanas līknes (atbilstoši šā pielikuma 268.1.8.apakšpunktam).
270. No viena parauga ar 0,15 % (m/m) fluora saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,012 % (m/m) vērtību4.
22. Dzīvsudraborganisko savienojumu pierādīšana un noteikšana
271. Ar dzīvsudraborganisko savienojumu pierādīšanas un noteikšanas metodi pierāda un nosaka šādus dzīvsudraborganiskos atvasinājumus, ko izmanto kā konservantus acu kosmētikas līdzekļos: tiomersāli (INN), nātrija 2-(etildzīvsudraba) benzoātu un fenildzīvsudrabu un tā sāļus.
22.1. Dzīvsudraborganisko savienojumu pierādīšana
272. Dzīvsudraborganiskie savienojumi ir kompleksā ar 1,5-difenil-3-tiokarbazonu. Kad ditizonāts ir ekstrahēts ar tetrahloroglekli, veic silikagela plānslāņa hromatogrāfiju. Ditizonātu plankumi parādās oranži.
273. Dzīvsudraborganisko savienojumu pierādīšanas metodei izmanto šādus reaģentus1:
273.1. Sērskābe, 25 % (v/v).
273.2. 1,5-difenil-3-tiokarbazons (ditizons): 0,8 mg 100 mililitros tetrahloroglekļa (atbilstoši šā pielikuma 273.4.apakšpunktam).
273.3. Slāpeklis.
273.4. Tetrahlorogleklis.
273.5. Attīstīšanas šķīdinātājs: heksāns/acetons 90:10 (v/v).
273.6. Standartšķīdums, 0,001 % ūdenī: nātrija 2-(etildzīvsudrabtio)benzoāta, etildzīvsudraba hlorīda vai metildzīvsudraba hlorīda, fenildzīvsudraba nitrāta vai fenildzīvsudraba acetāta, dzīvsudraba dihlorīda vai dzīvsudraba diacetāta.
273.7. Lietošanai gatavas silikagela plates (tai skaitā Merck 5721 vai līdzvērtīgas).
273.8. Nātrija hlorīds.
274. Dzīvsudraborganisko savienojumu pierādīšanas metodei izmanto šādu aprīkojumu:
274.1. Laboratorijas standartaprīkojums.
274.2. Plānslāņa hromatogrāfijas standartaiekārta.
274.3. Fāžu atdalīšanas filtrs.
275. Dzīvsudraborganisko savienojumu pierādīšanu veic šādā kārtībā:
275.1. Ekstrakcija:
275.1.1. Centrifūgas mēģenē atšķaida 1 g parauga, titrējot ar 20 ml destilēta ūdens. Iegūst maksimālo dispersiju un sasilda līdz 60 °C ūdens vannā. Pievieno 4 g nātrija hlorīda. Sakrata. Ļauj atdzist.
275.1.2. Centrifugē vismaz 20 minūtes ar 4500 apgriezieniem minūtē, lai no šķidruma atdalītu lielāko daļu cietvielas. Izfiltrē dalāmajā piltuvē un pievieno 0,25 ml sērskābes šķīduma (atbilstoši šā pielikuma 273.1.apakšpunktam).
275.1.3. Ekstrahē vairākas reizes ar 2 vai 3 ml ditizona šķīduma (atbilstoši šā pielikuma 273.2.apakšpunktam), līdz pēdējā organiskā fāze paliek zaļa.
275.1.4. Katru organisko fāzi pēc kārtas filtrē caur fāžu atdalīšanas filtru.
275.1.5. Ietvaicē slāpekļa atmosfērā.
275.1.6. Izšķīdina 0,5 ml tetrahloroglekļa. Šo šķīdumu lieto tūlīt saskaņā ar šī pielikuma 275.2.1.apakšpunktu.
275.2. Atdalīšana un pierādīšana:
275.2.1. 50 μl tetrahloroglekļa šķīduma, kas iegūts saskaņā ar šī pielikuma 275.1.6.apakšpunktu, tūlīt klāj uz silikagela plates. Vienlaicīgi saskaņā ar šī pielikuma 275.1.apakšpunktu apstrādā 10 ml standartšķīduma (atbilstoši šā pielikuma 273.6.apakšpunktam) un uz tās pašas plates klāj 50 μl šķīduma, kas iegūts saskaņā ar šā pielikuma 275.1.6.apakšpunktam.
275.2.2. Liek plati šķīdinātājā (atbilstoši šā pielikuma 273.5.apakšpunktam) un ļauj attīstīties 150 mm. Dzīvsudraborganiskie savienojumi parādās noturīgi iekrāsotu plankumu veidā, ja plati tūlīt pēc šķīdinātāja iztvaicēšanas pārsedz ar stiklu.
Piemēram, iegūst šādas Rf vērtības:
|
Rf |
Krāsa |
|
|
Tiomersāls |
0,33 |
Oranža |
|
Etildzīvsudraba hlorīds |
0,29 |
Oranža |
|
Metildzīvsudraba hlorīds |
0,29 |
Oranža |
|
Fenildzīvsudraba sāļi |
0,21 |
Oranža |
|
Dzīvsudraba(II) sāļi |
0,10 |
Oranža |
|
Dzīvsudraba diacetāts |
0,10 |
Oranža |
|
1,5-difenil-3-tiokarbazons |
0,09 |
Rozā |
22.2. Dzīvsudraborganisko savienojumu noteikšana*
276. Ar dzīvsudraborganisko savienojumu noteikšanas metodi noteikto dzīvsudraborganisko savienojumu saturu paraugā izsaka paraugā esošā dzīvsudraba masas procentos (m/m).
277. Dzīvsudraborganisko savienojumu noteikšanas metodes būtība ir kopējā dzīvsudraba mērīšana. Tāpēc vispirms jāpārliecinās, vai paraugā nav neorganiska dzīvsudraba, un jāpierāda dzīvsudraborganiskais atvasinājums paraugā. Pēc mineralizācijas atbrīvoto dzīvsudrabu mēra, veicot bezliesmas atomu absorbciju.
278. Dzīvsudraborganisko savienojumu noteikšanas metodei izmanto šādus reaģentus1:
278.1. Koncentrēta slāpekļskābe, ![]() = 1,41 g/ml.
= 1,41 g/ml.
278.2. Koncentrēta sērskābe, ![]() = 1,84 g/ml.
= 1,84 g/ml.
278.3. Ūdens bidestilāts.
278.4. Kālija permanganāts, 7 % (m/v) šķīdums.
278.5. Hidroksilamonija hlorīds, 1,5 % (m/v) šķīdums.
278.6. Dikālija peroksodisulfāts, 5 % (m/v) šķīdums.
278.7. Alvas dihlorīds, 10 % (m/v) šķīdums.
278.8. Koncentrēta sālsskābe, ![]() = 1,18 g/ml.
= 1,18 g/ml.
278.9. Ar palādija dihlorīdu piesūcināta stikla vate, 1 % (m/m).
279. Dzīvsudraborganisko savienojumu noteikšanas metodei izmanto šādu aprīkojumu:
279.1. Laboratorijas standartaprīkojums.
279.2. Iekārta, tai skaitā stikla trauki, kas nepieciešami dzīvsudraba noteikšanai, izmantojot bezliesmas atomu absorbciju (aukstā tvaika paņēmiens). Kivetes garums vismaz 100 mm.
280. Ievērojot visus mikrodzīvsudraba analīzes standarta drošības pasākumus, dzīvsudraborganisko savienojumu noteikšanu veic šādā kārtībā:
280.1. Precīzi nosver 150 mg parauga (m). Pievieno 10 ml slāpekļskābes (atbilstoši šā pielikuma 278.1.apakšpunktam) un ļauj reaģēt trīs stundas hermētiskā kolbā ūdens vannā 55 °C, regulāri sakratot. Vienlaicīgi izdara reaģentu tukšo pārbaudi.
280.2. Pēc atdzesēšanas pievieno 10 ml sērskābes (atbilstoši šā pielikuma 278.2.apakšpunktam) un vēlreiz liek ūdens vannā 55 °C uz 30 minūtēm.
280.3. Ieliek kolbu ledus vannā un uzmanīgi pievieno 20 ml ūdens (atbilstoši šā pielikuma 278.3.apakšpunktam).
280.4. Pievieno 2 ml 7 % kālija permanganāta šķīduma alikvotās daļas, līdz šķīdums iekrāsojas. Ieliek atpakaļ ūdens vannā 55 °C vēl uz 15 minūtēm.
280.5. Pievieno 4 ml dikālija peroksodisulfāta šķīduma (atbilstoši šā pielikuma 278.6.apakšpunktam). Turpina 30 minūtes sildīt ūdens vannā 55 °C.
280.6. Ļauj atdzist un pārnes kolbas saturu uz 100 ml mērkolbu. Izskalo kolbu ar 5 ml hidroksilamonija hlorīda 1,5 % šķīdumu un pēc tam četras reizes izskalo ar 10 ml ūdens bidestilāta. Šķīdumam būtu jābūt pilnīgi bezkrāsainam. Uzpilda līdz zīmei ar ūdens bidestilātu.
280.7. Liek 10 ml analizējamā šķīduma (atbilstoši šā pielikuma 280.6.apakšpunktam) stikla traukā dzīvsudraba noteikšanai ar aukstā tvaika paņēmienu (atbilstoši šā pielikuma 279.2.apakšpunktam). Atšķaida ar 100 ml ūdens bidestilāta, pēc tam ar 5 ml 25 % sērskābes un 5 ml 10 % alvas dihlorīda šķīduma. Pēc katras pievienošanas sajauc. Nogaida 30 sekundes, lai visi dzīvsudraba joni reducējas līdz metāliskam dzīvsudrabam, un veic mērījumu (n).
280.8. Ieliek ar palādija dihlorīdu piesūcinātu stikla vati starp dzīvsudraba reducēšanas trauku un kiveti (atbilstoši šā pielikuma 379.2.apakšpunktam). Atkārto šā pielikuma 280.7.apakšpunktā minēto darbību un reģistrē mērījumu. Ja mērījuma rezultāts nav nulle, mineralizācija nav bijusi pabeigta un analīze jāatkārto.
281. Dzīvsudraba daudzumu, kas izteikts dzīvsudraba masas procentos, aprēķina pēc formulas:
% dzīvsudraba = ![]() , kur
, kur
m – parauga masa (miligramos);
n – izmērītais dzīvsudraba daudzums (μg).
282. No viena parauga ar aptuveni 0,007 % dzīvsudraba koncentrāciju divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,00035 % vērtību4.
* Piezīmes.
Lai uzlabotu mineralizāciju, var būt nepieciešams sākt ar parauga atšķaidīšanu.
Ja ir aizdomas, ka substrāts absorbē dzīvsudrabu, noteikšanu veic ar standarta pievienošanas metodi.
23. Sārmu un sārmzemju sulfīdu noteikšana
283. Ar sārmu un sārmzemju sulfīdu metodi nosaka sulfīdus kosmētikas līdzekļos. Tioli vai citi reducējoši aģenti (tai skaitā sulfīti) netraucē.
284. Sulfīdu koncentrāciju izsaka sēra masas procentos.
285. Pēc vides paskābināšanas hidrogēnsulfīdu aizvada ar slāpekļa plūsmu, pēc tam nofiksē kadmija sulfīda veidā. Pēdējo izfiltrē un izskalo, un pēc tam nosaka jodometriski.
286. Sārmu un sārmzemju sulfīdu noteikšanas metodei izmanto šādus reaģentus1:
286.1. Koncentrēta sālsskābe, ![]() = 1,19 g/ml.
= 1,19 g/ml.
286.2. Nātrija tiosulfāts, 0,1 M standartšķīdums.
286.3. Jods, 0,05 M standartšķīdums.
286.4. Dinātrija sulfīds.
286.5. Kadmija diacetāts.
286.6. Koncentrēts amonjaks, ![]() = 0,90 g/ml.
= 0,90 g/ml.
286.7. Amonjakāls kadmija diacetāta šķīdums: izšķīdina 10 g kadmija diacetāta aptuveni 50 ml ūdens. Pievieno amonjaku (atbilstoši šā pielikuma 286.6.apakšpunktam), līdz nogulsnes izšķīst (tas ir aptuveni 20 ml). Uzpilda līdz 100 ml zīmei ar ūdeni.
286.8. Slāpeklis.
286.9. Amonjaka M šķīdums.
287. Sārmu un sārmzemju sulfīdu noteikšanas metodei izmanto šādu aprīkojumu:
287.1. Laboratorijas standartaprīkojums.
287.2. 100 ml trijkaklu apaļkolba ar pieslīpētiem kakliem.
287.3. Divas 150 ml koniskas kolbas ar pieslīpētiem kakliem, kas aprīkotas ar ierīci, kurai ir ievadcaurule un novadcaurule gāzes izlaišanai.
287.4. Viena piltuve ar garu kātu.
288. Sārmu un sārmzemju sulfīdu noteikšanu veic šādā kārtībā:
288.1. Sulfīdu atdalīšana:
288.1.1. Ņem iepakojumu, kas iepriekš nav bijis atvērts. Precīzi iesver apaļkolbā (atbilstoši šā pielikuma 287.2.apakšpunktam) kosmētikas līdzekļa masu (m) (gramos), kas atbilst ne vairāk kā 30 mg sulfīda jonu. Pievieno 60 ml ūdens un divus pilienus pretputu šķidruma.
288.1.2. Pārnes 50 ml šķīduma (atbilstoši šā pielikuma 286.7.apakšpunktam) uz abām koniskajām kolbām (atbilstoši šā pielikuma 287.3.apakšpunktam).
288.1.3. Apaļkolbu (atbilstoši šā pielikuma 287.2.apakšpunktam) aprīko ar piltuvi pilināšanai, ievadcauruli un novadcauruli. Novadcauruli pievieno pie koniskajām kolbām (atbilstoši šā pielikuma 287.3.apakšpunktam), kas savienotas ar teflona caurulēm.
Atdalīšanas iekārtas hermētiskumu pārbauda šādi: imitējot testa apstākļus, aizstāj nosakāmo kosmētikas līdzekli ar 10 ml sulfīda šķīduma (gatavots no šā pielikuma 286.4.apakšpunktā minētā dinātrija sulfīda), kas satur "X" mg sulfīda (nosaka jodometriski). "Y" pieņem par sulfīda miligramu skaitu šīs operācijas beigās. Starpība starp daudzumu "X" un daudzumu "Y" nedrīkst pārsniegt 3 %.
288.1.4. Laiž cauri slāpekli 15 minūtes ar ātrumu divi burbuļi sekundē, lai izspiestu gaisu, kas ir apaļkolbā (atbilstoši šā pielikuma 287.2.apakšpunktam).
288.1.5. Karsē apaļkolbu līdz 85±5 °C.
288.1.6. Pārtrauc slāpekļa atmosfēru un pa pilienam pievieno 40 ml sālsskābes (atbilstoši šā pielikuma 286.1.apakšpunktam).
288.1.7. Slāpekļa atmosfēru atjauno, kad gandrīz visa skābe ir pārnesta, atstājot nelielu šķidruma slāni, lai novērstu hidrogēnsulfīda noplūdi.
288.1.8. Karsēšanu pārtrauc pēc 30 minūtēm. Ļauj kolbai (atbilstoši šā pielikuma 287.2.apakšpunktam) atdzist un turpina laist cauri slāpekļa atmosfēru vismaz pusotru stundu.
288.2. Titrēšana:
288.2.1. Izfiltrē kadmija sulfīdu caur piltuvi ar garu kātu.
288.2.2. Izskalo koniskās kolbas (atbilstoši šā pielikuma 288.3.apakšpunktam) ar amonjaka šķīdumu (atbilstoši šā pielikuma 286.9.apakšpunktam) un izlej uz filtra. Pēc tam izskalo ar destilētu ūdeni un ar šo ūdeni mazgā filtrā aizturētās nogulsnes.
288.2.3. Nogulšņu mazgāšanu pabeidz ar 100 ml ūdens.
288.2.4. Papīra filtru liek pirmajā koniskajā kolbā, kurā bija nogulsnes. Pievieno 25 ml (n1) joda šķīduma (atbilstoši šā pielikuma 286.3.apakšpunktam), aptuveni 20 ml sālsskābes (atbilstoši šā pielikuma 286.1.apakšpunktam) un 50 ml destilēta ūdens.
288.2.5. Nosaka joda pārākumu, izmantojot nātrija tiosulfāta šķīdumu (n2) (atbilstoši šā pielikuma 286.2.apakšpunktam).
289. Sulfīda saturu paraugā, izteiktu sēra masas procentos, aprēķina pēc šādas formulas:
% sēra = ![]() , kur
, kur
n1 – izlietotā joda standartšķīduma daudzums (mililitros);
x1 – šī šķīduma molaritāte;
n2 – nātrija tiosulfāta standartšķīduma daudzums (mililitros);
x2 – šī šķīduma molaritāte;
m – parauga masa (gramos).
290. No viena parauga ar aptuveni 2 % (m/m) sulfīda saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,2 % (m/m) vērtību4.
24. Glicerīna 1-(4-aminobenzoāta) pierādīšana un noteikšana
24.1. Glicerīna 1-(4-aminobenzoāta) pierādīšana
291. Ar glicerīna 1-(4-aminobenzoāta) pierādīšanas metodi pierāda alfa-monogliceril-4-aminobenzoātu (glicerīna 1-(4-aminobenzoātu)) un etil-4-aminobenzoātu (benzokaīnu INN), kas var būt kā piemaisījums.
292. Glicerīna 1-(4-aminobenzoātu) pierāda silikagela plānslāņa hromatogrāfijā ar fluorescējošu indikatoru, un brīvo pirmējo aminogrupu nosaka pēc diazokrāsvielas veidošanās uz plates.
293. Glicerīna 1-(4-aminobenzoāta) pierādīšanas metodei izmanto šādus reaģentus1:
293.1. Šķīdinātāja maisījums: cikloheksāns/propan-2-ols/stabilizēts dihlormetāns 48:64:9 (v/v/v).
293.2. Attīstīšanas šķīdinātājs: petrolētera (40-60)/benzola/acetona/amonija hidroksīda šķīdums (vismaz 25 % NH3) 35:35:35:1 (v/v/v/v).
293.3. Attīstīšanas šķīdinātājs:
293.3.1. nātrija nitrīts: 1 g 100 mililitros 1 M sālsskābes (gatavo tieši pirms lietošanas);
293.3.2. 2-naftols: 0,2 g 100 mililitros 1 M kālija hidroksīda.
293.4. Standartšķīdumi: alfa-monogliceril-4-aminobenzoāts: 0,05 g 100 mililitros šķīdinātāja maisījuma (293.1.apakšpunkts); etil-4-amino šī pielikuma benzoāts: 0,05 g 100 mililitros šķīdinātāja maisījuma (atbilstoši šā pielikuma 293.1.apakšpunktam).
293.5. Silikagela 60 F254 plates, 0,25 mm biezas, 200 × 200 mm.
294. Glicerīna 1-(4-aminobenzoāta) pierādīšanas metodei izmanto šādu aprīkojumu:
294.1. Plānslāņa hromatogrāfijas standartaprīkojums.
294.2. Ultraskaņas vanna.
294.3. Sīkporu filtrs FH, 0,5 μm vai līdzvērtīgs.
295. Glicerīna 1-(4-aminobenzoāta) pierādīšanu veic šādā kārtībā:
295.1. Parauga sagatavošana: iesver 1,5 g analizējamā kosmētikas līdzekļa 10 ml mērkolbā ar aizbāzni. Uzpilda līdz zīmei ar šķīdinātāju (atbilstoši šā pielikuma 293.1.apakšpunktam). Noslēdz un atstāj vienu stundu istabas temperatūrā ultraskaņas vannā. Izfiltrē caur sīkporu filtru (atbilstoši šā pielikuma 294.3.apakšpunktam) un filtrātu izmanto hromatogrāfijai.
295.2. Plānslāņa hromatogrāfija: uznes 10 ml parauga šķīduma (atbilstoši šā pielikuma 295.1.apakšpunktam) un katra standartšķīduma (atbilstoši šā pielikuma 293.4.apakšpunktam) uz plates (atbilstoši šā pielikuma 293.5.apakšpunktam). Attīsta hromatogrammu līdz 150 mm augstumam kamerā, kas piesātināta ar šķīdinātāju (atbilstoši šā pielikuma 293.2.apakšpunktam). Ļauj platei nožūt apkārtējās vides temperatūrā.
295.3. Attīstīšana:
295.3.1. Apskata plati 254 nm ultravioletā gaismā.
295.3.2. Pilnīgi nožuvušo plati apsmidzina ar šķīdumu (atbilstoši šā pielikuma 293.3.1.apakšpunktam). Ļauj žūt istabas temperatūrā 1 minūti un tūlīt apsmidzina ar šķīdinātāju (atbilstoši šā pielikuma 293.3.2.apakšpunktam). Žāvē plati žāvēšanas skapī 60 °C. Plankumi parādās oranži. Alfa-monogliceril-4-aminobenzoāta Rf = 0,07, etil-4-aminobenzoāta Rf = 0,55.
24.2. Glicerīna 1-(4-aminobenzoāta) noteikšana*
296. Ar glicerīna 1-(4-aminobenzoāta) noteikšanas metodi nosaka alfa-monogliceril-4-aminobenzoātu un etil-4-aminobenzoātu. Ar to nevar noteikt vairāk kā 5 % (m/m) alfa-monogliceril-4-aminobenzoāta un 1 % (m/m) etil-4-aminobenzoāta.
297. Alfa-monogliceril-4-aminobenzoāta un etil-4-aminobenzoāta saturu izsaka kosmētikas līdzekļa masas procentos (% m/m).
298. Analizējamo kosmētikas līdzekli suspendē metanolā un pēc attiecīgas parauga apstrādes nosaka augstas izšķirtspējas šķidruma hromatogrāfijā (turpmāk - HPLC).
299. Glicerīna 1-(4-aminobenzoāta) noteikšanas metodei izmanto šādus reaģentus1,2:
299.1. Metanols.
299.2. Kālija dihidrogēnortofosfāts (KH2PO4).
299.3. Cinka diacetāts (Zn(CH3COO)2·2H2O).
299.4. Etiķskābe (d![]() = 1,05).
= 1,05).
299.5. Kālija heksacianoferāts (K4 (Fe(CN)6)·3H2O).
299.6. Etil-4-hidroksibenzoāts.
299.7. Alfa-monogliceril-4-aminobenzoāts.
299.8. Etil-4-aminobenzoāts.
299.9. Fosfāta buferšķīdums (0,02 M): izšķīdina 2,72 g kālija dihidrogēnortofosfāta (atbilstoši šā pielikuma 299.2.apakšpunktam) vienā litrā ūdens.
299.10. Eluants: fosfāta buferšķīdums (atbilstoši šā pielikuma 298.9.apakšpunktam)/metanols 61:39 (v/v). Kustīgās fāzes sastāvu var mainīt, lai sasniegtu izšķirtspēju ar koeficientu
R = 1,5 ![]() , kur
, kur
R1 un R2 – pīķu izdalīšanas laiki (minūtēs);
W1 un W2 – pīķu platumi pusaugstumā (milimetros);
d' – pašrakstītāja ātrums (milimetros minūtē).
299.11. Alfa-monogliceril-4-aminobenzoāta standartšķīdums: precīzi nosver aptuveni 40 mg alfa-monogliceril-4-aminobenzoāta un ievada 100 ml mērkolbā. Izšķīdina 40 mililitros metanola. Uzpilda līdz zīmei ar buferšķīdumu (atbilstoši šā pielikuma 299.9.apakšpunktam) un sajauc.
299.12. Etil-4-aminobenzoāta standartšķīdums: precīzi nosver aptuveni 40 mg etil-4-aminobenzoāta un ievada 100 ml mērkolbā. Izšķīdina 40 mililitros metanola. Uzpilda līdz zīmei ar buferšķīdumu (atbilstoši šā pielikuma 299.9.apakšpunktam) un sajauc.
299.13. Iekšējā standarta šķīdums: precīzi nosver aptuveni 50 mg etil-4-hidroksibenzoāta, pārnes uz 100 ml mērkolbu, izšķīdina 40 mililitros metanola, uzpilda līdz zīmei ar buferšķīdumu (atbilstoši šā pielikuma 298.9.apakšpunktam) un sajauc.
299.14. Standartšķīdumi: gatavo četrus standartšķīdumus, izšķīdinot 100 mililitros eluanta (atbilstoši šā pielikuma 298.10.apakšpunktam) saskaņā ar šo tabulu:
|
Standartšķīdums |
Alfa-monogliceril-4-aminobenzoāts |
Etil-4-aminobenzoāts |
Etil-4-hidroksibenzoāts |
|||
|
(μg/ml)* |
ml (299.11.) |
(μg/ml)* |
ml (299.11.) |
(μg/ml)* |
ml (299.11.) |
|
|
I |
8 |
2 |
8 |
2 |
50 |
10 |
|
II |
16 |
4 |
12 |
3 |
50 |
10 |
|
III |
24 |
6 |
16 |
4 |
50 |
10 |
|
IV |
40 |
10 |
20 |
5 |
50 |
10 |
|
* Šīs vērtības ir norādītas un atbilst precīzajai masai (atbilstoši šā pielikuma 299.11., 299.12. un 299.13.apakšpunktam). Šos šķīdumus var gatavot citādi. |
||||||
299.15. Karesa I šķīdums: izšķīdina 26,5 g kālija heksacianoferāta ūdenī un uzpilda līdz 250 ml.
299.16. Karesa II šķīdums: izšķīdina 54,9 g cinka diacetāta un 7,5 ml etiķskābes (atbilstoši šā pielikuma 298.4.apakšpunktam) ūdenī un uzpilda līdz 250 ml.
299.17. Merck Lichrosorb RP-18 vai līdzvērtīgs ar daļiņu vidējo izmēru 5 μm.
300. Glicerīna 1-(4-aminobenzoāta) noteikšanas metodei izmanto šādu aprīkojumu:
300.1. Laboratorijas standartaprīkojums.
300.2. Augstas izšķirtspējas hromatogrāfijas iekārta ar maināma garuma ultravioleto staru detektoru un kolonnas termostatu, kas noregulēts uz 45 °C.
300.3. Nerūsējoša tērauda kolonna: garums 250 mm, iekšējais diametrs 4,6 mm, pildījums Lichrosorb RP-18 (atbilstoši šā pielikuma 299.17.apakšpunktam).
300.4. Ultraskaņas vanna.
301. Glicerīna 1-(4-aminobenzoāta) noteikšanu veic šādā kārtībā:
301.1. Parauga gatavošana:
301.1.1. Precīzi iesver aptuveni 1 g parauga 100 ml vārglāzē un pievieno 10 ml metanola.
301.1.2. Liek vārglāzi ultraskaņas vannā uz 20 minūtēm, lai rodas suspensija. Šādi iegūto suspensiju kvantitatīvi pārnes uz 100 ml mērkolbu ar ne vairāk kā 75 ml eluanta (atbilstoši šā pielikuma 299.10.apakšpunktam).
Secīgi pievieno 1 ml Karesa I šķīduma (atbilstoši šā pielikuma 299.15.apakšpunktam) un 1 ml Karesa II šķīduma (atbilstoši šā pielikuma 299.16.apakšpunktam) un sajauc pēc katras pievienošanas. Uzpilda līdz zīmei ar eluantu (atbilstoši šā pielikuma 299.10.apakšpunktam), vēlreiz sajauc un izfiltrē caur kroku filtrpapīru.
301.1.3. Ar pipeti pārnes 3,0 ml saskaņā ar šī pielikuma 301.1.2.apakšpunktu iegūtā filtrāta un 5,0 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 299.13.apakšpunktam) uz 50 ml mērkolbu. Uzpilda līdz zīmei ar eluantu (atbilstoši šā pielikuma 299.10.apakšpunktam) un sajauc. Šādi iegūto šķīdumu izmanto hromatogrāfiskajā analīzē (atbilstoši šā pielikuma 301.2.apakšpunktam).
301.2. Hromatogrāfija:
301.2.1. Kustīgās fāzes (atbilstoši šā pielikuma 299.10.apakšpunktam) plūsmas ātrumu noregulē uz 1,2 ml/min un kolonnas temperatūru uz 45 °C.
301.2.2. Detektora viļņu garumu (atbilstoši šā pielikuma 300.2.apakšpunktam) noregulē uz 274 nm.
301.2.3. Ar mikrošļirci vismaz divas reizes pa 20 μl šķīduma (atbilstoši šā pielikuma 301.1.3.apakšpunktam) ievada hromatogrāfā un izmēra pīķu laukumus.
301.3. Kalibrēšanas līkne:
301.3.1. Ievada 20 ml no katra standartšķīduma (atbilstoši šā pielikuma 299.14.apakšpunktam) un izmēra pīķu laukumus.
301.3.2. Katrai koncentrācijai aprēķina alfa-monogliceril-4-aminobenzoāta pīķu un iekšējā standarta pīķu laukumu attiecību. Šo attiecību atzīmē uz abscisu ass un attiecīgo masu attiecību uz ordinātu ass.
301.3.3. Tādu pašu procedūru piemēro etil-4-hidroksibenzoātam.
302. Aprēķinus veic šādā kārtībā:
302.1. No kalibrēšanas līknes, kas iegūta saskaņā ar 301.3.apakšpunktu, nolasa masu attiecības (RP1, RP2), kuras atbilst saskaņā ar 301.2.3.apakšpunktu aprēķināto pīķu laukumu attiecībām, kur
RP1 – alfa-monogliceril-4-aminobenzoāta masa/etil-4-hidroksibenzoāta masa;
RP2 – etil-4-aminobenzoāta masa/etil-4-hidroksibenzoāta masa.
302.2. No šādi iegūtajām masu attiecībām aprēķina alfa-monogliceril-4-aminobenzoāta un etil-4-aminobenzoāta saturu masas procentos (% m/m) pēc formulas:
Rp % (m/m) alfa-monogliceril-4-aminobenzoāta = ![]()
Rp % (m/m) etil-4-aminobenzoāta = ![]() , kur
, kur
q – etil-4-hidroksibenzoāta (iekšējā standarta) daudzums (miligramos) saskaņā ar šī pielikuma 299.12.apakšpunktu;
p – parauga daudzums (gramos) saskaņā ar šā pielikuma 301.1.1.apakšpunktu.
303. No viena parauga ar 5 % (m/m) alfa-monogliceril-4-aminobenzoāta saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,25 %4.
304. No viena parauga ar 1 % (m/m) etil-4-aminobenzoāta saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,10 %4.
* Piezīmes.
Pirms analīzes pārbauda, vai paraugā ir vielas, kuru pīķi hromatogrammā var pārklāties ar iekšējā standarta (etil-4aminobenzoāta) pīķi.
Lai pārbaudītu, vai nav nekādu traucējumu, noteikšanu atkārto, mainot metanola proporciju kustīgajā fāzē relatīvi par 10 %.
25. Hlorbutanola noteikšana
305. Ar hlorbutanola noteikšanas metodi nosaka hlorbutanolu (INN) līdz maksimālajai koncentrācijai 0,5 % (m/m) jebkurā kosmētikas līdzeklī, izņemot aerosolus.
306. Hlorbutanola saturu izsaka kosmētikas līdzekļa masas procentos (% m/m).
307. Kad analizējamais kosmētikas līdzeklis ir attiecīgi apstrādāts, noteikšanu veic gāzu hromatogrāfijā, par iekšējo standartu izmantojot 2,2,2-trihloretanolu.
308. Hlorbutanola noteikšanas metodei izmanto šādus reaģentus1:
308.1. Hlorbutanol (1,1,1-trihlor-2-metilpropan-2-ols).
308.2. 2,2,2-trihloretanols.
308.3. Absolūtais spirts (etanols).
308.4. Hlorbutanola standartšķīdums: 0,025 g 100 mililitros etanola (m/v).
308.5. Standartšķīdums: 4 mg 2,2,2-trihloretanols 100 mililitros etanola (m/v).
309. Hlorbutanola noteikšanas metodei izmanto šādu aprīkojumu:
309.1. Laboratorijas standartaprīkojums.
309.2. Gāzu hromatogrāfs ar elektronu detektoru, Ni 63.
310. Hlorbutanola noteikšanu veic šādā kārtībā:
310.1. Parauga gatavošana: precīzi nosver 0,1–0,3 g (p g) parauga. Liek 100 ml mērkolbā. Izšķīdina etanolā, pievieno 1 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 308.5.apakšpunktam) un uzpilda līdz zīmei ar etanolu.
310.2. Gāzu hromatogrāfijas apstākļi:
310.2.1. Ekspluatācijas nosacījumiem jānodrošina izšķirtspējas koeficients R = 1,5.
![]() , kur
, kur
R1 un R2 – pīķu izdalīšanas laiki (minūtēs);
W1 un W2 – pīķu platumi pusaugstumā (milimetros);
d' – pašrakstītāja ātrums (milimetros minūtē).
310.2.2. Piemēram, šādi darba apstākļi nodrošina vajadzīgo izšķirtspēju:
|
Kolonna |
I |
II |
|
Materiāls |
Stikls |
Nerūsējošs tērauds |
|
Garums |
1,80 m |
3 m |
|
Diametrs |
3 mm |
3 mm |
|
Nekustīgā fāze |
10 % Carbowax 20 M TPA uz Gaschrom Q, ar daļiņu izmēru 80–100 |
5 % OV 17 uz Chromosorb WAW DMCS, ar daļiņu izmēru 80–100 |
|
Paraugu kondicionēšana |
2–3 dienas 190 °C |
|
|
Temperatūra: |
||
|
inžektors |
150 °C |
|
|
kolonna |
150 °C |
100 °C |
|
detektors |
200 °C |
150 °C |
|
Nesējgāze |
Slāpeklis |
Argons/metāns 95:5 (v/v) |
|
Plūsmas ātrums |
35 ml/min |
35 ml/min |
310.3. Standarta līkne. Lietojot piecas 100 ml mērkolbas, pievieno 1 ml standartšķīduma (atbilstoši šā pielikuma 308.5.apakšpunktam) un attiecīgi 0,2, 0,3, 0,4, 0,5 un 0,6 ml šķīduma (atbilstoši šā pielikuma 308.4.apakšpunktam), uzpilda līdz zīmei ar etanolu un sajauc. Ievada 1 μl katra šķīduma hromatogrāfā saskaņā ar darba apstākļiem, kas minēti šā pielikuma 310.2.2.apakšpunktā, un konstruē kalibrēšanas līkni, uz abscisu ass atzīmējot hlorbutanola masas attiecību pret 2,2,2-trihloretanola masu un uz ordinātu ass – attiecīgo pīķu laukumu attiecību.
310.4. Ievada 1 μl saskaņā ar šā pielikuma 310.1.apakšpunktu iegūtā šķīduma un turpina darbību saskaņā ar apstākļiem, kas minēti šī pielikuma 310.2.2.apakšpunktā.
311. Aprēķinus veic šādā kārtībā:
311.1. Pēc standarta līknes (atbilstoši šā pielikuma 310.3.apakšpunktam) aprēķina daudzumu a, ko izsaka hlorbutanola miligramos šķīdumā (atbilstoši šā pielikuma 310.1.apakšpunktam).
311.2. Hlorbutanola saturu paraugā aprēķina pēc formulas:
% (m/m) hlorbutanola =
![]()
312. No viena parauga ar 0,5 % (m/m) hlorbutanola saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,01 %4. Ja rezultāts ir vienāds ar maksimālo atļauto koncentrāciju vai pārsniedz to, jāpārbauda, vai nav pārklāšanās.
26. Hinīna pierādīšana un noteikšana
26.1. Hinīna pierādīšana
313. Ar hinīna pierādīšanas metodi nosaka hinīnu šampūnos un matu losjonos.
314. Pierāda silikagela plānslāņa hromatogrāfijā. Hinīnu nosaka pēc tā zilās fluorescences skābā vidē 360 nm. Turpmākajam apstiprinājumam fluorescenci var novērst ar broma tvaikiem, amonjaka tvaiki izraisīs dzeltenīgu fluorescenci.
315. Hinīna pierādīšanas metodei izmanto šādus reaģentus1:
315.1. Silikagela plates bez fluorescences indikatoriem, 0,25 mm biezas, 200 × 200 mm.
315.2. Attīstīšanas šķīdinātājs: toluols/dietilēteris/dihlormetāns/dietilamīns 20:20:20:8 (v/v/v/v).
315.3. Metanols.
315.4. Sērskābe (96 %; ![]() = 1,84).
= 1,84).
315.5. Dietilēteris.
315.6. Attīstīšanas aģents: atdzesētā traukā 95 mililitriem dietilētera uzmanīgi pievieno 5 ml sērskābes (atbilstoši šā pielikuma 315.4.apakšpunktam).
315.7. Broms.
315.8. Amonija hidroksīda šķīdums (28 %; ![]() = 0,90).
= 0,90).
315.9. Hinīns, bezūdens.
315.10. Standartšķīdums: precīzi iesver aptuveni 100,0 mg bezūdens hinīna mērkolbā un izšķīdina 100 mililitros metanola.
316. Hinīna pierādīšanas metodei izmanto šādu aprīkojumu:
316.1. Standarta iekārta plānslāņa hromatogrāfijai.
316.2. Ultraskaņas vanna.
316.3. Sīkporu filtrs, FH 0,5 μm vai līdzvērtīgs ar piemērotu filtrēšanas iekārtu.
317. Hinīna pierādīšanu veic šādā kārtībā:
317.1. Parauga gatavošana: precīzi iesver 100 ml mērkolbā tādu parauga daudzumu, kurā var būt aptuveni 100 mg hinīna, izšķīdina un uzpilda līdz zīmei ar metanolu. Noslēdz kolbu un atstāj vienu stundu istabas temperatūrā ultraskaņas vannā. Izfiltrē (atbilstoši šā pielikuma 316.3.apakšpunktam) un filtrātu lieto hromatogrāfijā.
317.2. Plānslāņa hromatogrāfija: uznes 1,0 ml standartšķīduma (atbilstoši šā pielikuma 315.10.apakšpunktam) un 1,0 ml parauga šķīduma (atbilstoši šā pielikuma 317.1.apakšpunktam) uz silikagela plates (atbilstoši šā pielikuma 315.1.apakšpunktam). Attīsta hromatogrammu 150 mm garumā ar šķīdinātāju (atbilstoši šā pielikuma 315.2.apakšpunktam) kamerā, kas iepriekš piesātināta ar šķīdinātāju (atbilstoši šā pielikuma 315.2.apakšpunktam).
317.3. Attīstīšana:
317.3.1. Nožāvē plati istabas temperatūrā.
317.3.2. Apsmidzina ar reaģentu (atbilstoši šā pielikuma 315.6.apakšpunktam).
317.3.3. Ļauj platei žūt vienu stundu istabas temperatūrā.
317.3.4. Apskata ultravioletās lampas gaismā, kuras viļņu garums ir 360 nm. Hinīna plankums fluorescē spilgti zils.
Tabulā ir galveno hinīnam radniecīgo alkaloīdu Rf vērtību piemēri, kas attiecas uz attīstīšanu ar šķīdinātāju (atbilstoši šā pielikuma 315.2.apakšpunktam).
|
Alkaloīds |
RF |
|
Hinīns |
0,20 |
|
Hinidīns |
0,29 |
|
Cinhonīns |
0,33 |
|
Cinhonidīns |
0,27 |
|
Hidrohinidīns |
0,17 |
317.3.5. Turpmākam hinīna klātbūtnes apstiprinājumam paraugā plati apmēram vienu stundu apstrādā ar broma tvaiku. Fluorescence zūd. Ja to pašu plati apstrādā ar amonjaka tvaiku (atbilstoši šā pielikuma 315.8.apakšpunktam), plankumi no jauna parādās brūnā krāsā un, ja plati atkal apskata 360 nm ultravioletā gaismā, var novērot dzeltenīgu fluorescenci. Noteikšanas robeža: 0,1 μg hinīna.
26.2. Hinīna noteikšana
318. Ar hinīna noteikšanas metodi nosaka hinīnu. Ar to var noteikt maksimālo atļauto 0,5 % (m/m) koncentrāciju šampūnos un 0,2 % koncentrāciju matu losjonos.
319. Ar hinīna noteikšanas metodi noteiktā hinīna saturu izsaka kosmētikas līdzekļa masas procentos (% m/m).
320. Kad analizējamais kosmētikas līdzeklis ir attiecīgi apstrādāts, noteikšanu veic augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC).
321. Hinīna noteikšanas metodei izmanto šādus reaģentus1, 2:
321.1. Acetonitrils.
321.2. Kālija dihidrogēnortofosfāts (KH2PO4).
321.3. Ortofosforskābe (85 %; ![]() = 1,7).
= 1,7).
321.4. Tetrametilamonija bromīds.
321.5. Hinīns, bezūdens.
321.6. Metanols.
321.7. Ortofosforskābes šķīdums (0,1 M): iesver 11,53 g ortofosforskābes (atbilstoši šā pielikuma 321.3.apakšpunktam) mērkolbā un izšķīdina 1000 ml ūdens.
321.8. Kālija dihidrogēnortofosfāta šķīdums (0,1 M): iesver 13,6 g kālija dihidrogēnortofosfāta (atbilstoši šā pielikuma 321.2.apakšpunktam) mērkolbā un izšķīdina 1000 ml ūdens.
321.9. Tetrametilamonija bromīda šķīdums: mērkolbā izšķīdina 15,40 g tetrametilamonija bromīda 1000 ml ūdens.
321.10. Eluants: ortofosforskābe (atbilstoši šā pielikuma 321.7.apakšpunktam)/kālija dihidrogēnortofosfāts (atbilstoši šā pielikuma 321.8.apakšpunktam)/tetrametilamonija bromīds (atbilstoši šā pielikuma 321.9.apakšpunktam)/
ūdens/acetonitrils 10:50:100:340:90 (v/v/v/v/v).
Kustīgās fāzes sastāvu var mainīt, lai sasniegtu izšķirtspējas koeficientu R = 1,5.
![]() , kur
, kur
R1 un R2 – pīķu izdalīšanas laiki (minūtēs);
W1 un W2 – pīķu platumi pusaugstumā (milimetros);
d' – pašrakstītāja ātrums (milimetros minūtē).
321.11. Silīcija oksīds apstrādāts ar oktadecilsilānu, 10 μm.
321.12. Standartšķīdumi: precīzi iesver attiecīgi aptuveni 5,0, 10,0, 15,0 un 20,0 mg bezūdens hinīna 100 ml kolbās. Uzpilda līdz zīmei ar metanolu un krata, līdz hinīns izšķīst. Visus paraugus izfiltrē caur 0,5 mm filtru.
322. Hinīna noteikšanas metodei izmanto šādu aprīkojumu:
322.1. Laboratorijas standartaprīkojums.
322.2. Ultraskaņas vanna.
322.3. Augstas izšķirtspējas šķidruma hromatogrāfijas iekārta ar maināma viļņu garuma detektoru.
322.4. Kolonna: garums 250 mm, iekšējais diametrs 4,6 mm, pildījums – silīcija oksīds (atbilstoši šā pielikuma 321.11.apakšpunktam).
322.5. Sīkporu filtrs, FH 0,5 mm vai līdzvērtīgs piemērotai filtrēšanas iekārtai.
323. Hinīna noteikšanu veic šādā kārtībā:
323.1. Parauga gatavošana: precīzi iesver 100 ml mērkolbā tādu daudzumu kosmētikas līdzekļa, kurā ir vismaz 10,0 mg bezūdens hinīna, pievieno 20 ml metanola un liek kolbu ultraskaņas vannā uz 20 minūtēm. Uzpilda līdz zīmei ar metanolu. Samaisa šķīdumu un pēc tam izfiltrē alikvoto daļu (atbilstoši šā pielikuma 322.5.apakšpunktam).
323.2. Hromatogrāfija:
323.2.1. Plūsmas ātrums: 1,0 ml/min.
323.2.2. Detektora viļņu garums: 332 nm.
323.2.3. Ievadītais tilpums: 10 µl filtrētā šķīduma (atbilstoši šā pielikuma 323.1.apakšpunktam).
323.2.4. Mērījums: pīķu laukums.
323.3. Kalibrēšanas līkne: vismaz trīs reizes ievada 10,0 ml no katra standartšķīduma (atbilstoši šā pielikuma 321.12.apakšpunktam), izmēra pīķu laukumu un aprēķina vidējo laukumu atbilstīgi katrai koncentrācijai. Izveido kalibrēšanas līkni un pārliecinās, vai tā ir taisna.
324. Aprēķinus veic šādā kārtībā:
324.1. Pēc kalibrēšanas līknes (atbilstoši šā pielikuma 323.3.apakšpunktam) nosaka bezūdens hinīna daudzumu miligramos ievadītajā tilpumā (atbilstoši šā pielikuma 323.2.apakšpunktam).
324.2. Bezūdens hinīna koncentrāciju paraugā masas procentos (% m/m) nosaka pēc šādas formulas:
% (m/m) bezūdens hinīna = ![]() , kur
, kur
B – filtrētā šķīduma (atbilstoši šā pielikuma 323.1.apakšpunktam) 10 mikrolitros noteiktais bezūdens hinīna daudzums (mikrogramos);
A – parauga masa (gramos) (atbilstoši šā pielikuma 323.1.apakšpunktam).
325. No viena parauga ar 0,5 % (m/m) bezūdens hinīna saturu divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt 0,02 %. No viena parauga ar 0,2 % (m/m) bezūdens hinīna saturu divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt 0,01 %4.
27. Neorganisko sulfītu un hidrogēnsulfītu pierādīšana un noteikšana
326. Ar neorganisko sulfītu un hidrogēnsulfītu pierādīšanas un noteikšanas metodi pierāda un nosaka neorganiskos sulfītus un hidrogēnsulfītus kosmētikas līdzekļos, kuru sastāvā ir ūdens vai spirta fāze un līdz 0,2 % sēra dioksīda koncentrācija.
27.1. Neorganisko sulfītu un hidrogēnsulfītu pierādīšana
327. Paraugu karsē sālsskābē un atbrīvoto sēra dioksīdu pierāda pēc smaržas vai ar indikatorpapīru.
328. Neorganisko sulfītu un hidrogēnsulfītu pierādīšanas metodei izmanto šādus reaģentus1:
328.1. Sālsskābe (4 M).
328.2. Kālija jodāta cietes papīrs vai līdzvērtīgs.
329. Neorganisko sulfītu un hidrogēnsulfītu pierādīšanas metodei izmanto šādu aprīkojumu:
329.1. Laboratorijas standartaprīkojums.
329.2. Kolba (25 ml) ar īsu atteces dzesinātāju.
330. Neorganisko sulfītu un hidrogēnsulfītu pierādīšanu veic šādā kārtībā:
330.1. Liek kolbā (atbilstoši šā pielikuma 329.2.apakšpunktam) aptuveni 2,5 g parauga un pievieno 10 ml sālsskābes (atbilstoši šā pielikuma 328.1.apakšpunktam).
330.2. Sajauc un karsē līdz viršanai.
330.3. Pārbauda, vai izdalās sērs.
27.2. Neorganisko sulfītu un hidrogēnsulfītu noteikšana
331. Sulfīta vai hidrogēnsulfīta saturu paraugā izsaka sēra dioksīda masas procentos.
332. Pēc parauga paskābināšanas atbrīvoto sēra dioksīdu iedestilē ūdeņraža peroksīda šķīdumā. Izveidojušos sērskābi titrē ar standartizētu nātrija hidroksīda šķīdumu.
333. Neorganisko sulfītu un hidrogēnsulfītu noteikšanas metodei izmanto šādus reaģentus1:
333.1. Ūdeņraža peroksīds, 0,2 % (m/v) (katru dienu gatavo jaunu).
333.2. Ortofosforskābe (85 %; ![]() = 1,75).
= 1,75).
333.3. Metanols.
333.4. Nātrija hidroksīds, (0,01 M) standartšķīdums.
333.5. Slāpeklis.
333.6. Indikators: maisījums 1:1 (v/v) metilsarkanā (0,03 % m/v etanolā) un metilēnzilā (0,05 % m/v etanolā). Izfiltrē šķīdumu.
334. Neorganisko sulfītu un hidrogēnsulfītu noteikšanas metodei izmanto šādu aprīkojumu:
334.1. Laboratorijas standartaprīkojums.
334.2. Destilācijas iekārta (7.attēls).
7.attēls
Tenera (Tanner) sēra dioksīda destilācijas iekārta. Visi
izmēri milimetros
335. Neorganisko sulfītu un hidrogēnsulfītu noteikšanu veic šādā kārtībā:
335.1. Precīzi iesver aptuveni 2,5 g parauga destilācijas kolbā A (7.attēls).
335.2. Pievieno 60 ml ūdens un 50 ml metanola un sajauc.
335.3. Destilāta uztvērējā D (7.attēls) iepilina 10 ml ūdeņraža peroksīda (atbilstoši šā pielikuma 333.1.apakšpunktam), 60 ml ūdens un dažus pilienus indikatora (atbilstoši šā pielikuma 333.6.apakšpunktam). Pievieno dažus pilienus nātrija hidroksīda (atbilstoši šā pielikuma 333.4.apakšpunktam), līdz indikators kļūst zaļš.
335.4. Atkārto šī pielikuma 335.3.apakšpunktā minēto darbību attiecībā uz skalotni E (7.attēls).
335.5. Samontē iekārtu un noregulē slāpekļa plūsmu aptuveni uz 60 burbuļiem minūtē.
335.6. Ielaiž 15 ml ortofosforskābes (atbilstoši šā pielikuma 333.2.apakšpunktam) no piltuves destilācijas kolbā A.
335.7. Strauji sakarsē līdz viršanai un pēc tam lēni silda, kopā 30 minūtes.
335.8. Atvieno destilāta uztvērēju D. Izskalo cauruli un pēc tam titrē ar nātrija hidroksīda šķīdumu (atbilstoši šā pielikuma 333.4.apakšpunktam), līdz indikators kļūst zaļš (atbilstoši šā pielikuma 333.6.apakšpunktam).
336. Aprēķina sulfīta vai hidrogēnsulfīta saturu paraugā pēc masas, izmantojot formulu:
% m/m sēra dioksīda =
![]() , kur
, kur
M – nātrija hidroksīda šķīduma (atbilstoši šā pielikuma 333.4.apakšpunktam) molārā koncentrācija;
V – titrēšanai (atbilstoši šā pielikuma 335.8.apakšpunktam) vajadzīgā nātrija hidroksīda (atbilstoši šā pielikuma 333.4.apakšpunktam) tilpums (mililitros);
m – parauga (atbilstoši šā pielikuma 335.1.apakšpunktam) masa (gramos).
337. No viena parauga ar 0,2 % (m/m) sēra dioksīda saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,006 % dioksīda pēc smaržas vai indikatora papīra (atbilstoši šā pielikuma 328.2.apakšpunktam) 4.
28. Sārmu metālu hlorātu pierādīšana un noteikšana
338. Ar sārmu metālu hlorātu pierādīšanas un noteikšanas metodi pierāda un nosaka hlorātus zobu pastās un citos kosmētikas līdzekļos.
28.1. Sārmu metālu hlorātu pierādīšana
339. Hlorātus atdala no citiem halogenātiem plānslāņa hromatogrāfijā un pierāda ar jodīda oksidāciju, kuras rezultātā veidojas jods.
340. Sārmu metālu hlorātu pierādīšanas metodei izmanto šādus reaģentus1:
340.1. Standartšķīdumi: svaigi sagatavoti kālija hlorāta, bromāta un jodāta ūdens šķīdumi 0,2 % (m/v).
340.2. Attīstīšanas šķīdinātājs: amonjaka šķīdums 28 % (m/v)/acetons/butanols 60:130:30 (v/v/v).
340.3. Kālija jodīda ūdens šķīdums 5 % (m/v).
340.4. Cietes šķīdums 1–5 % (m/v).
340.5. Sālsskābe (1 M).
340.6. Lietošanai gatavas ar celulozi pārklātas plānslāņa plates (0,25 mm).
341. Sārmu metālu hlorātu pierādīšanas metodei izmanto standarta iekārtu plānslāņa hromatogrāfijai.
342. Sārmu metālu hlorātu pierādīšanu veic šādā kārtībā:
342.1. Apmēram 1 g parauga ekstrahē ar ūdeni, izfiltrē un atšķaida līdz 25 ml.
342.2. Uz plates (atbilstoši šā pielikuma 340.6.apakšpunktam) uznes 2 ml šķīduma (atbilstoši šā pielikuma 342.1.apakšpunktam) un 2 ml alikvotu no katra no trijiem standartšķīdumiem (atbilstoši šā pielikuma 340.1.apakšpunktam).
342.3. Liek plati kamerā un augšupejošā hromotogrāfijā attīsta apmēram trīs ceturtdaļas plates garuma ar šķīdinātāju (atbilstoši šā pielikuma 340.2.apakšpunktam).
342.4. Izņem no kameras un ļauj šķīdinātājam iztvaikot. Iztvaikošana var aizņemt līdz pat divām stundām.
342.5. Apsmidzina plati ar kālija jodīdu (atbilstoši šā pielikuma 340.3.apakšpunktam) un ļauj žūt apmēram piecas minūtes.
342.6. Apsmidzina plati ar cietes šķīdumu (atbilstoši šā pielikuma 340.4.apakšpunktam) un ļauj žūt apmēram piecas minūtes.
342.7. Apsmidzina plati ar sālsskābi (atbilstoši šā pielikuma 340.5.apakšpunktam).
343. Ja paraugā ir hlorāts, pēc pusstundas parādās zils (iespējams, brūns) plankums ar Rf vērtību aptuveni 0,7–0,8.
|
Halogenāti |
RF |
|
Jodāts |
0–0,2 |
|
Bromāts |
0,5–0,6 |
|
Hlorāts |
0,7–0,8 |
Bromāti un jodāti reaģē uzreiz. Bromātu un hlorātu plankumus nesajauc.
28.2. Sārmu metālu hlorātu noteikšana
344. Hlorāta daudzumu paraugā izsaka hlorāta masas procentos.
345. Hlorātu skābā vidē reducē ar cinka pulveri. Izveidojušos hlorīdu nosaka, potenciometriski titrējot ar sudraba nitrāta šķīdumu. Līdzīgi pirms reducēšanas nosaka iespējamos halogenīdus.
346. Sārmu metālu hlorātu noteikšanas metodei izmanto šādus reaģentus1:
346.1. Etiķskābe, 80 % (m/m).
346.2. Cinka pulveris.
346.3. Sudraba nitrāta standartšķīdums (0,1 M).
347. Sārmu metālu hlorātu noteikšanas metodei izmanto šādu aprīkojumu:
347.1. Laboratorijas standartaprīkojums.
347.2. Potenciometrs ar sudraba indikatora elektrodu.
348. Sārmu metālu hlorātu noteikšanu veic šādā kārtībā:
348.1. Parauga gatavošana: precīzi iesver aptuveni 2 g daudzumu m centrifūgas mēģenē. Pievieno apmēram 15 ml etiķskābes un rūpīgi sajauc. Pagaida 30 minūtes un centrifugē 15 minūtes ar ātrumu 2000 apgr./min. Pārnes centrifugātu uz 50 ml mērkolbu. Centrifugēšanu atkārto divas reizes, atlikumam pievienojot 15 ml etiķskābes. Šķīdumu, kurā ir hlorāts, savāc tajā pašā mērkolbā. Uzpilda līdz zīmei ar etiķskābi.
348.2. Hlorāta reducēšana: ņem 20 ml šķīduma (atbilstoši šā pielikuma 348.1.apakšpunktam) un pievieno 0,6 g cinka pulvera. Uzvāra kolbā, kas aprīkota ar atteces dzesinātāju. Pēc 30 minūšu vārīšanas atdzesē un izfiltrē. Izskalo kolbu ar ūdeni. Izfiltrē un filtrātu apvieno ar saskalām.
348.3. Hlorīda noteikšana: titrē 20 ml šķīduma (atbilstoši šā pielikuma 348.2.apakšpunktam) ar sudraba nitrātu (atbilstoši šā pielikuma 346.3.apakšpunktam), lietojot potenciometru (atbilstoši šā pielikuma 347.2.apakšpunktam). Tāpat ar sudraba nitrātu titrē 20 ml šķīduma (atbilstoši šā pielikuma 348.1.apakšpunktam).
Ja kosmētikas līdzeklī ir broma vai joda atvasinājumi, kas pēc reducēšanas var atbrīvot bromīdus vai jodīdus, titrēšanas līknē ir vairāki kāpuma punkti. Šādā gadījumā titrētā šķīduma (atbilstoši šā pielikuma 346.3.apakšpunktam) tilpums, kas atbilst hlorīdam, ir starpība starp pēdējo un priekšpēdējo kāpuma punktu.
349. Hlorāta saturu paraugā (% m/m) aprēķina pēc formulas:
% (m/m) hlorāta (Clo3-) = ![]() , kur
, kur
V – šķīduma (atbilstoši šā pielikuma 348.2.apakšpunktam) titrēšanā izlietotā sudraba nitrāta (atbilstoši šā pielikuma 346.3.apakšpunktam) tilpums (mililitros);
V' – šķīduma (atbilstoši šā pielikuma 348.1.apakšpunktam) 20 mililitru titrēšanā izlietotā sudraba nitrāta (atbilstoši šā pielikuma 346.3.apakšpunktam) tilpums (mililitros);
M – sudraba nitrāta standartšķīduma (atbilstoši šā pielikuma 346.3.apakšpunktam) molaritāte;
m – parauga masa (gramos).
350. No viena parauga ar 3–5 % (m/m) hlorāta saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,07 %4.
29. Nātrija jodāta pierādīšana un noteikšana
351. Ar nātrija jodāta pierādīšanas un noteikšanas metodi pierāda un nosaka nātrija jodātu kosmētikas līdzekļu saskalās.
29.1. Nātrija jodāta pierādīšana
352. Nātrija jodātu atdala no citiem halogenātiem plānslāņa hromatogrāfijā un pierāda ar jodīda oksidāciju, kuras rezultātā veidojas jods.
353. Nātrija jodāta pierādīšanas metodei izmanto šādus reaģentus1:
353.1. Standartšķīdumi: svaigi sagatavots kālija hlorāta, bromāta un jodāta ūdens šķīdums 0,01 % (m/v).
353.2. Attīstīšanas šķīdinātājs. Amonjaka šķīdums 28 % (m/v)/acetons/butanols 60:130:30 (v/v/v).
353.3. Kālija jodīda ūdens šķīdums 5 % (m/v).
353.4. Cietes šķīdums 1–5 % (m/v).
353.5. Sālsskābe (1 M).
354. Nātrija jodāta pierādīšanas metodei izmanto šādu aprīkojumu:
354.1. Lietošanai gatavas celulozes plānslāņa hromatogrāfijas plates (0,25 mm).
354.2. Plānslāņa hromatogrāfijas standartiekārta.
355. Nātrija jodāta pierādīšanu veic šādā kārtībā:
355.1. Apmēram 1 g parauga ekstrahē ar ūdeni, izfiltrē un atšķaida apmēram līdz 10 ml.
355.2. Uznes 2 μl šī šķīduma un 2 μl alikvotu no katra no trijiem standartšķīdumiem (atbilstoši šā pielikuma 353.1.apakšpunktam) uz plates (atbilstoši šā pielikuma 354.1.apakšpunktam) starta līnijas.
355.3. Liek plati kamerā un augšupejošā hromatogrāfijā attīsta apmēram trīs ceturtdaļas plates garuma ar šķīdinātāju (atbilstoši šā pielikuma 353.2.apakšpunktam).
355.4. Izņem plati no kameras un ļauj šķīdinātājam iztvaikot apkārtējās vides temperatūrā. Iztvaikošana var ilgt līdz divām stundām.
355.5. Apsmidzina plati ar kālija jodīdu (atbilstoši šā pielikuma 353.3.apakšpunktam) un ļauj žūt apmēram piecas minūtes.
355.6. Apsmidzina plati ar cietes šķīdumu (atbilstoši šā pielikuma 353.4.apakšpunktam) un ļauj žūt apmēram piecas minūtes.
355.7. Beidzot apsmidzina ar sālsskābi (atbilstoši šā pielikuma 353.5.apakšpunktam).
356. Ja paraugā ir jodāts, tūlīt parādās zils (iespējams, brūns vai arī pēc laika brūnējošs) plankums ar Rf vērtību aptuveni 0–0,2. Jāatzīmē, ka bromāti reaģē uzreiz, ja Rf vērtība ir aptuveni 0,5–0,6, un hlorāti – apmēram pēc 30 minūtēm, ja Rf vērtība ir attiecīgi 0,7–0,8.
29.2. Nātrija jodāta noteikšana
357. Nātrija jodāta saturu izsaka nātrija jodāta masas procentos.
358. Nātrija jodātu izšķīdina ūdenī un nosaka augstas izšķirtspējas šķidruma hromatogrāfijā, secīgi izmantojot apgrieztās fāzes C18 kolonnu un anjonu apmaiņas kolonnu.
359. Nātrija jodāta noteikšanas metodei izmanto šādus reaģentus1, 2:
359.1. Sālsskābe (4 M).
359.2. Nātrija sulfīta ūdens šķīdums, 5 % (m/v).
359.3. Nātrija jodāta standartšķīdums. Gatavo standartšķīdumu, kas satur 50 mg nātrija jodāta uz 100 ml ūdens.
359.4. Kālija dihidrogēnortofosfāts.
359.5. Dinātrija hidrogēnortofosfāts; 2H2O.
359.6. HPLC kustīgā fāze: izšķīdina 3,88 g kālija dihidrogēnortofosfāta un 1,19 g dinātrija hidrogēnortofosfāta 2H2O (atbilstoši šā pielikuma 359.5.apakšpunktam) 1 litrā ūdens.
Iegūtā šķīduma pH ir 6,2.
359.7. Universālais indikatorpapīrs, pH 1–11.
360. Nātrija jodāta noteikšanas metodei izmanto šādu aprīkojumu:
360.1. Laboratorijas standartaprīkojums.
360.2. Apaļš papīra filtrs ar diametru 110 mm, Schleicher and Schuell Nr.575 vai līdzvērtīgs.
360.3. Augstas izšķirtspējas šķidruma hromatogrāfs ar maināma viļņu garuma detektoru.
360.4. Kolonnas: garums 120 mm, iekšējais diametrs 4,6 mm, skaits – divas secīgi savienotas daļas: pirmā – Necleosil R 5 C18 vai līdzvērtīga kolonna, otrā – Vydac TM-301-SB vai līdzvērtīga kolonna.
361. Nātrija jodāta noteikšanu veic šādā kārtībā:
361.1. Parauga gatavošana:
361.1.1. Šķidrie paraugi (šampūni):
361.1.1.1. Precīzi iesver apmēram 1,0 g analizējamā parauga 10 ml graduētā mēģenē vai mērkolbā, kam ir stikla aizbāznis.
361.1.1.2. Uzpilda ar ūdeni līdz zīmei un sajauc.
361.1.1.3. Ja nepieciešams, izfiltrē.
361.1.1.4. Veicot HPLC, nosaka jodātu šķīdumā saskaņā ar šī pielikuma 361.2.apakšpunktu.
361.1.2. Cietie paraugi (ziepes):
361.1.2.1. Sasmalcina daļu parauga un precīzi iesver apmēram 1,0 g analizējamā daudzuma 100 ml mērcilindrā, kam ir stikla aizbāznis.
361.1.2.2. Uzpilda līdz 50 ml ar ūdeni un stipri krata vienu minūti.
361.1.2.3. Centrifugē un izfiltrē caur filtrpapīru vai ļauj maisījumam nostāties vismaz vienu nakti.
361.1.2.4. Stipri sakrata želejveida šķīdumu un izfiltrē caur filtrpapīru.
361.1.2.5. Veicot HPLC, nosaka jodātu filtrātā saskaņā ar šī pielikuma 362.2.apakšpunktu.
361.2. Hromatogrāfija:
361.2.1. Plūsmas ātrums: 1 ml/min.
361.2.2. Detektora viļņu garums: 210 nm.
361.2.3. Ievadītais tilpums: 10 ml.
361.2.4. Mērījums: pīķu laukums.
361.3. Kalibrēšana:
361.3.1. Ar pipeti attiecīgi iepilina 1,0, 2,0, 5,0, 10,0 un 20,0 ml nātrija jodāta standartšķīduma (atbilstoši šā pielikuma 359.3.apakšpunktam) 50 ml mērkolbās.
361.3.2. Uzpilda līdz zīmei un sajauc.
361.3.3. Šādi iegūtajos šķīdumos ir attiecīgi 0,01, 0,02, 0,05, 0,10 un 0,20 mg nātrija jodāta uz vienu mililitru.
361.3.4. Ievada 10 μl devu katra standarta jodāta šķīduma šķidruma hromatogrāfā un hromatografē.
361.3.5. Nosaka jodāta pīķu laukumu un konstruē līkni, attiecinot pīķa laukumu pret nātrija jodāta koncentrāciju.
362. Aprēķina nātrija jodāta saturu masas procentos (% m/m) pēc formulas:
% (m/m) nātrija jodāta = ![]() , kur
, kur
m – analizējamā parauga daudzuma (atbilstoši šā pielikuma 361.1.apakšpunktam) masa (gramos);
V – tā parauga šķīduma kopējais tilpums (mililitros), ko iegūst saskaņā ar šā pielikuma 361.1.apakšpunktu;
c – nātrija jodāta koncentrācija (miligramos uz mililitru), ko nosaka pēc kalibrēšanas līknes (atbilstoši šā pielikuma 361.3.apakšpunktam).
363. No viena parauga ar 0,1 % (m/m) nātrija jodāta saturu divās paralēlās noteikšanās iegūto rezultātu starpība nedrīkst pārsniegt 0,002 %4.
364. Apstiprinājumu nosaka šādi:
364.1. Princips: paskābinātā kosmētikas līdzekļa šķīdumā ar sulfītu jodātu (IO3) reducē par jodīdu (I) un iegūto šķīdumu izpēta HPLC. Ja pīķis, kura izdalīšanas laiks atbilst jodāta izdalīšanas laikam, pazūd pēc apstrādes ar sulfītu, sākotnējo pīķi visticamāk var attiecināt uz jodātu.
364.2. Procedūra:
364.2.1. Ar pipeti koniskajā kolbā iepilina 5 ml parauga šķīduma, ko iegūst saskaņā ar šī pielikuma 361.1.apakšpunktu.
364.2.2. Ar sālsskābi (atbilstoši šā pielikuma 359.1.apakšpunktam) noregulē šķīduma pH uz 3 vai zemāk. Pārbauda ar universālo indikatorpapīru.
364.2.3. Pievieno trīs pilienus nātrija sulfīta šķīduma (atbilstoši šā pielikuma 359.2.apakšpunktam) un sajauc.
364.2.4. Ievada 10 μl šķīduma šķidruma hromatogrāfā.
364.2.5. Salīdzina šo hromatogrammu ar hromatogrammu, kas ar to pašu paraugu iegūta saskaņā ar šā pielikuma 361.apakšpunktu.
30. Sudraba nitrāta pierādīšana un noteikšana kosmētikas līdzekļos
30.1. Sudraba nitrāta pierādīšana kosmētikas līdzekļos
365. Ar sudraba nitrāta pierādīšanas metodi pierāda sudraba nitrātu kā sudrabu kosmētikas līdzekļos uz ūdens bāzes.
366. Sudrabu pierāda raksturīgas baltas nogulsnes, kas veidojas ar hlorīda joniem.
367. Sudraba nitrāta pierādīšanas metodei izmanto šādus reaģentus1:
367.1. Sālsskābes šķīdums, 2 M.
367.2. Amonjaka šķīdums: koncentrētu amonija hidroksīda šķīdumu (d20 = 0,88 g/ml) atšķaida ar tādu pašu daudzumu ūdens un sajauc.
367.3. Slāpekļskābes šķīdums, 2 M.
368. Sudraba nitrāta pierādīšanas metodei izmanto šādu aprīkojumu.
368.1. Laboratorijas standarta aprīkojums.
368.2. Centrifūga.
369. Sudraba nitrāta pierādīšanu veic šādā kārtībā:
369.1. Apmēram 1 gramam parauga centrifūgas mēģenē pa pilienam pievieno 2 M sālsskābes šķīdumu, līdz beidzas izgulsnēšanās, sajauc un centrifugē.
369.2. Nolej centrifugātu un nogulsnes vienreiz skalo ar pieciem pilieniem auksta ūdens. Nolej mazgājumus.
369.3. Centrifūgas mēģenē nogulsnēm pielej nedaudz ūdens. Karsē līdz viršanai un maisa.
369.4. Centrifugē karstu, nolej centrifugātu.
369.5. Nogulsnēm pievieno dažus pilienus amonjaka šķīduma (atbilstoši šā pielikuma 367.2.apakšpunktam), sajauc un centrifugē.
369.6. Vienam pilienam centrifugāta uz priekšmetstikliņa pievieno dažus pilienus 2 M slāpekļskābes šķīduma.
369.7. Baltas nogulsnes liecina par sudraba klātbūtni.
30.2. Sudraba nitrāta noteikšana kosmētikas līdzekļos
370. Ar sudraba nitrāta noteikšanas metodi nosaka sudraba nitrātu kā sudrabu kosmētikas līdzekļos, kas paredzēti skropstu vai uzacu krāsošanai.
371. Sudrabu produktā nosaka, veicot atomu absorbcijas spektrometriju.
372. Sudraba nitrāta noteikšanas metodei izmanto šādus reaģentus1:
372.1. Slāpekļskābes šķīdums, 0,02 M.
372.2. Sudraba standartšķīdumi:
372.2.1. Rezerves sudraba standartšķīdums, 1000 µg/ml 0,5 M slāpekļskābes šķīdumā (SpectrosoL vai līdzvērtīgā).
372.2.2. Sudraba standartšķīdums, 100 µg/ml: ar pipeti 10 ml sudraba standartšķīduma (atbilstoši šā pielikuma 372.2.1.apakšpunktam) pārnes uz 100 ml mērkolbu. Uzpilda līdz atzīmei ar 0,02 M slāpekļskābes šķīdumu un sajauc. Šo standartšķīdumu izmanto svaigu un glabā tumšā stikla pudelē.
373. Sudraba nitrāta noteikšanas metodei izmanto šādu aprīkojumu:
373.1. Laboratorijas standarta aprīkojums.
373.2. Atomu absorbcijas spektrofotometrs, kas aprīkots ar doba sudraba katoda lampu.
374. Sudraba nitrāta noteikšanu veic šādā kārtībā:
374.1. Parauga sagatavošana:
374.1.1. Precīzi nosver aptuveni 0,1 g (m gramu) vienmērīga produkta parauga.
374.1.2. Kvantitatīvi pārnes uz viena litra mērkolbu, uzpilda līdz atzīmei ar 0,02 M slāpekļskābes šķīdumu un sajauc.
374.2. Atomu absorbcijas spektrometrijas apstākļi:
374.2.1. Liesma: gaisa–acetilēna.
374.2.2. Viļņu garums: 338,3 nm.
374.2.3. Fona korekcija: vajadzīga.
374.2.4. Degšanas apstākļi: nabadzīga liesma; maksimālai absorbcijai – optimāls degļa garums un degšanas apstākļi.
374.3. Kalibrēšana:
374.3.1. Ar pipeti vairākās 100 ml mērkolbās iepilda attiecīgi 1,0, 2,0, 3,0, 4,0, un 5,0 ml sudraba standartšķīduma (atbilstoši šā pielikuma 372.2.2.apakšpunktam). Katru kolbu uzpilda līdz atzīmei ar 0,02 M slāpekļskābes šķīdumu un sajauc. Šie šķīdumi satur attiecīgi 1,0, 2,0, 3,0, 4,0 un 5,0 mg sudraba uz mililitru.
374.3.2. Izmēra 0,02 M slāpekļskābes šķīduma absorbciju un iegūto vērtību kalibrēšanas līknē izmanto kā sudraba nulles koncentrāciju. Izmēra katra sudraba kalibrēšanas standarta (atbilstoši šā pielikuma 374.3.1.apakšpunktam) absorbciju. Uzzīmē kalibrēšanas līkni, attiecinot absorbcijas vērtības pret sudraba koncentrāciju.
374.4. Noteikšana: izmēra paraugšķīduma (atbilstoši šā pielikuma 374.1.apakšpunktam) absorbciju. No kalibrēšanas līknes nolasa sudraba koncentrāciju, kas atbilst paraugšķīduma absorbcijas vērtībai.
375. Aprēķina sudraba nitrāta saturu paraugā masas procentos (% m/m) pēc formulas:
% (m/m) sudraba nitrāta = ![]() , kur
, kur
m – analizējamā parauga (atbilstoši šā pielikuma 374.1.apakšpunktam) masa (gramos);
c – sudraba koncentrācija paraugšķīdumā (atbilstoši šā pielikuma 374.1.apakšpunktam) (mikrogramos uz mililitru), ko nosaka pēc kalibrēšanas līknes.
376. No viena parauga ar 4 % (m/m) sudraba nitrāta saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,05 % (m/m) 4.
31. Selēna disulfīda pierādīšana un noteikšana pretblaugznu šampūnos
31.1. Selēna disulfīda pierādīšana pretblaugznu šampūnos
377. Ar selēna disulfīda pierādīšanas metodi pierāda selēna disulfīdu kā selēnu pretblaugznu šampūnos.
378. Selēnu pierāda raksturīga dzeltenīgi oranža krāsa, kas rodas, reaģējot ar urīnvielu un kālija jodīdu.
379. Selēna disulfīda pierādīšanas metodei izmanto šādus reaģentus1:
379.1. Koncentrēta (d20 = 1,42 g/ml) slāpekļskābe.
379.2. Urīnviela.
379.3. Kālija jodīda šķīdums, 10 % (m/v): 100 ml ūdens izšķīdina 10 g kālija jodīda.
380. Selēna disulfīda pierādīšanas metodei izmanto šādu aprīkojumu:
380.1. Laboratorijas standarta aprīkojums.
380.2. Reakcijas kamera, kuras tilpums ir 100 ml.
380.3. Reaktors ar apsildīšanas bloku.
380.4. Filtrpapīrs (vatmaņpapīrs Nr.42 vai līdzvērtīgs) vai 0,45 µm membrānfiltrs.
381. Selēna disulfīda pierādīšanu veic šādā kārtībā:
381.1. Reakcijas kamerā aptuveni 1 g šampūna pievieno 2,5 ml koncentrētas slāpekļskābes (atbilstoši šā pielikuma 381.1.apakšpunktam) un ļauj reaģēt 30 minūtes aptuveni 150 °C reaktorā ar apsildīšanas bloku.
381.2. Izreaģējušo paraugu atšķaida līdz 25 ml ar ūdeni un izfiltrē caur filtrpapīru vai 0,45 mm membrānfiltru.
381.3. Filtrāta daudzumam, kas vienāds ar 2,5 ml, pievieno 5 ml ūdens, 2,5 ml urīnvielas un vāra. Atdzesē un pievieno 1 ml kālija jodīda šķīduma (atbilstoši šā pielikuma 379.3.apakšpunktam).
381.4. Dzelteni oranža krāsa, kas strauji kļūst tumšāka, liecina par selēna klātbūtni.
31.2. Selēna disulfīda noteikšana pretblaugznu šampūnos
382. Ar selēna disulfīda noteikšanas metodi nosaka selēna disulfīdu kā selēnu pretblaugznu šampūnos, kuros selēna disulfīda saturs ir līdz 4,5 % (m/m).
383. Paraugam ļauj reaģēt ar slāpekļskābi un reakcijas produktā selēnu nosaka, veicot atomu absorbcijas spektrometriju.
384. Selēna disulfīda noteikšanas metodei izmanto šādus reaģentus1:
384.1. Koncentrēta (d20 = 1,42 g/ml) slāpekļskābe.
384.2. Slāpekļskābes šķīdums, 5 % (v/v): 500 ml ūdens vārglāzē, nepārtaukti maisot, pievieno 50 ml koncentrētas slāpekļskābes. Šo šķīdumu pārnes uz vienlitra mērkolbu un uzpilda līdz atzīmei.
384.3. Selēna rezerves standartšķīdums, 1000 mg/ml 0,5 M slāpekļskābes šķīdumā (SpectrosoL vai līdzvērtīgā).
385. Selēna disulfīda noteikšanas metodei izmanto šādu aprīkojumu.
385.1. Laboratorijas standarta aprīkojums.
385.2. Reakcijas kamera, kuras tilpums ir 100 ml.
385.3. Reaktors ar apsildīšanas bloku.
385.4. Filtrpapīrs (vatmaņpapīrs Nr.42 vai līdzvērtīgs) vai 0,45 mm membrānfiltrs.
385.5. Atomu absorbcijas spektrofotometrs, kas aprīkots ar doba selēna katoda lampu.
386. Selēna disulfīda noteikšanu veic šādā kārtībā:
386.1. Parauga sagatavošana:
386.1.1. Reakcijas kamerā precīzi iesver aptuveni 0,2 g (m gramus) homogēna šampūna parauga.
386.1.2. Pievieno 5 ml koncentrētas slāpekļskābes un ļauj 150 ºC temperatūrā reaģēt vienu stundu reaktorā ar apsildīšanas bloku.
386.1.3. Šķīdumam ļauj atdzist un atšķaida līdz 100 ml ar ūdeni. Izfiltrē caur filtrpapīru vai 0,45 mm membrānfiltru un izfiltrēto šķīdumu saglabā noteikšanai.
386.2. Atomu absorbcijas spektrometrijas apstākļi:
386.2.1. Liesma: gaisa–acetilēna;
386.2.2. Viļņu garums: 196,0 nm;
386.2.3. Fona korekcija: vajadzīga;
386.2.4. Degšanas apstākļi: nabadzīga liesma; maksimālai absorbcijai – optimāls degļa garums un degšanas apstākļi.
386.3. Kalibrēšana:
386.3.1. Ar pipeti vairākās 100 ml mērkolbās iepilda attiecīgi 1,0, 2,0, 3,0, 4,0, un 5,0 ml selēna standartšķīduma (atbilstoši šā pielikuma 383.3.apakšpunktam). Katru kolbu uzpilda līdz atzīmei ar 5 % (v/v) slāpekļskābes šķīdumu un sajauc. Šie šķīdumi satur attiecīgi 10, 20, 30, 40 un 50 mg selēna uz mililitru.
386.3.2. Izmēra 5 % slāpekļskābes šķīduma absorbciju un iegūto vērtību kalibrēšanas līknē izmanto kā selēna nulles koncentrāciju. Izmēra katra selēna kalibrēšanas standarta (atbilstoši šā pielikuma 386.3.1.apakšpunktam) absorbciju. Uzzīmē kalibrēšanas līkni, attiecinot absorbcijas vērtības pret selēna koncentrāciju.
386.4. Noteikšana:
386.4.1. Izmēra paraugšķīduma (atbilstoši šā pielikuma 386.1.3.apakšpunktam) absorbciju.
386.4.2. No kalibrēšanas līknes nolasa selēna koncentrāciju, kas atbilst parauga šķīduma absorbcijas vērtībai.
387. Aprēķina selēna disulfīda saturu paraugā masas procentos (% m/m), izmantojot formulu:
% (m/m) selēna disulfīda = ![]() , kur
, kur
m – analizējamā parauga (atbilstoši šā pielikuma 386.1.1.apakšpunktam) masa (gramos);
c – selēna koncentrācija paraugšķīdumā (atbilstoši šā pielikuma 386.1.3.apakšpunktam) (mikrogramos uz mililitru), ko nosaka pēc kalibrēšanas līknes.
388. No viena parauga ar 1 % (m/m) selēna disulfīda saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,05 % (m/m) 4.
32. Šķīstošā bārija un stroncija noteikšana pigmentos, kas ir sāļu un laku formā
32.1. Šķīstošā bārija noteikšana*
389. Ar šķīstošā bārija noteikšanas metodi veic šķīstošā bārija ekstrakciju un noteikšanu pigmentos sāļu un laku formā.
390. Pigmentu ekstrahē ar 0,07 M sālsskābes šķīdumu noteiktos apstākļos un bārija daudzumu ekstraktā nosaka, veicot atomu absorbcijas spektrometriju.
391. Šķīstošā bārija noteikšanas metodei izmanto šādus reaģentus1:
391.1. Absolūtais etanols.
391.2. Sālsskābes šķīdums, 0,07 M.
391.3. Sālsskābes šķīdums, 0,5 M.
391.4. Kālija hlorīda 8 % (m/v) šķīdums: 16 g kālija hlorīda izšķīdina 200 ml 0,07 M sālsskābes šķīdumā.
391.5. Bārija standartšķīdumi:
391.5.1. Bārija rezerves standartšķīdums, 1000 mg/ml 0,5 M slāpekļskābes šķīdumā (SpectrosoL vai līdzvērtīgā).
391.5.2. Bārija standartšķīdums, 100 mg/ml: ar pipeti 20,0 ml bārija standartšķīduma (atbilstoši šā pielikuma 391.5.1.apakšpunktam) pārnes uz 100 ml mērkolbu. Uzpilda līdz atzīmei ar 0,07 M slāpekļskābes šķīdumu un sajauc.
392. Šķīstošā bārija noteikšanas metodei izmanto šādu aprīkojumu:
392.1. Laboratorijas standartaprīkojums.
392.2. pH metrs ar ±0,02 vienību precizitāti.
392.3. Mehāniskais kratītājs.
392.4. Membrānfiltrs, kura poru lielums ir 0,45 mm.
392.5. Atomu absorbcijas spektrofotometrs, kas aprīkots ar doba bārija katoda lampu.
393. Šķīstošā bārija noteikšanu veic šādā kārtībā:
393.1. Parauga sagatavošana:
393.1.1. Koniskā kolbā precīzi iesver aptuveni 0,5 g (m gramus) pigmenta. Lai nodrošinātu pietiekamu tilpumu efektīvai kratīšanai, nelieto kolbas, kuru tilpums ir mazāks par 150 ml.
393.1.2. Ar pipetes palīdzību pievieno 1,0 ml etanola un pagroza kolbu tā, lai pigments pilnīgi saslapinās. No biretes pievieno precīzi tādu daudzumu 0,07 M sālsskābes šķīduma, kas nepieciešams, lai skābes tilpuma attiecība pret pigmenta masu ir precīzi 50 mililitri uz gramu. Kopējais ekstrahējošās vielas un etanola tilpums ir V ml. Kolbas saturu krata piecas sekundes, nodrošinot, lai sastāvdaļas labi sajaucas.
393.1.3. Ar pH metru izmēra iegūtās suspensijas pH, un, ja tas ir virs 1,5, pa pilienam pievieno 0,5 M sālsskābes šķīdumu, līdz sasniedz pH līmeni 1,4–1,5.
393.1.4. Kolbu noslēdz un 60 minūtes krata ar mehānisko kratītāju. Kratītājs jādarbina pietiekami ātri, lai rastos putas. Izfiltrē caur 0,45 mm membrānfiltru un savāc filtrātu. Ekstraktu necentrifugē pirms filtrēšanas. Ar pipeti 5,0 ml filtrāta pārnes uz 50 ml mērkolbu; uzpilda līdz atzīmei ar 0,07 M sālsskābes šķīdumu un sajauc. Šo šķīdumu lieto arī stroncija noteikšanai (atbilstoši šā pielikuma 32.2.apakšnodaļai).
393.1.5. Ar pipeti 5,0 ml kālija hlorīda šķīduma (atbilstoši šā pielikuma 391.4.apakšpunktam) un atšķaidītā filtrāta (atbilstoši šā pielikuma 393.1.4.apakšpunktam) alikvoto daļu (WBa ml) pārnes uz 100 ml mērkolbu, lai nodrošinātu koncentrāciju 3–10 mg bārija uz mililitru (sākumam būtu piemērota 10 ml alikvotā daļa). Uzpilda līdz atzīmei ar 0,07 M sālsskābes šķīdumu un sajauc.
393.1.6. Tajā pašā dienā, veicot atomu absorbcijas spektrometriju, nosaka bārija koncentrāciju šķīdumā (atbilstoši šā pielikuma 393.1.5.apakšpunktam).
393.2. Atomu absorbcijas spektrometrijas apstākļi:
393.2.1. Liesma: slāpekļa oksīda–acetilēna.
393.2.2. Fona korekcija: nav vajadzīga.
393.2.3. Degšanas apstākļi: nabadzīga liesma; maksimālai absorbcijai – optimāls degļa garums un degšanas apstākļi.
393.3. Kalibrēšana:
393.3.1. Ar pipeti vairākās 100 ml mērkolbās iepilda attiecīgi 1,0, 2,0, 3,0, 4,0, un 5,0 ml bārija standarta šķīduma (atbilstoši šā pielikuma 391.5.2.apakšpunktam). Uz katru kolbu ar pipeti pārnes 5,0 ml kālija hlorīda šķīduma (atbilstoši šā pielikuma 391.4.apakšpunktam); uzpilda līdz atzīmei ar 0,07 M sālsskābes šķīdumu un sajauc. Šie šķīdumi satur attiecīgi 2,0, 4,0, 6,0, 8,0 un 10,0 mg bārija uz mililitru. Līdzīgi sagatavo tukšu šķīdumu, bez bārija standarta šķīduma.
393.3.2. Izmēra tukšā šķīduma (atbilstoši šā pielikuma 393.3.1.apakšpunktam) absorbciju un iegūto vērtību kalibrēšanas līknē izmanto kā bārija nulles koncentrāciju. Izmēra katra bārija kalibrēšanas standarta (atbilstoši šā pielikuma 393.3.1.apakšpunktam) absorbciju. Uzzīmē kalibrēšanas līkni, attiecinot absorbcijas vērtības pret bārija koncentrāciju.
393.4. Noteikšana: izmēra parauga šķīduma (atbilstoši šā pielikuma 393.1.5.apakšpunktam) absorbciju. No kalibrēšanas līknes nolasa bārija koncentrāciju, kas atbilst parauga šķīduma absorbcijas vērtībai.
394. Šķīstošā bārija saturu (% m/m) pigmentā izsaka pēc formulas:
% (m/m) šķīstošā bārija = ![]() , kur
, kur
m – analizējamā parauga (atbilstoši šā pielikuma 393.1.1.apakšpunktam) masa (gramos);
c – šķīstošā bārija koncentrācija parauga šķīdumā (atbilstoši šā pielikuma 393.1.5.apakšpunktam) (mikrogramos uz mililitru), ko nosaka pēc kalibrēšanas līknes;
V – ekstrahējošās vielas kopējais tilpums (mililitros) (atbilstoši šā pielikuma 393.1.2.apakšpunktam);
WBa – ekstrakta tilpums (mililitros) (atbilstoši šā pielikuma 393.1.5.apakšpunktam).
395. Labākā šīs metodes atkārtojamības prognoze, ja šķīstošā bārija saturs ir 2 % (m/m), ir 0,3 %4.
*Piezīmes.
1. Konkrētos apstākļos bārija absorbciju var uzlabot ar kalciju. To var neitralizēt, pievienojot magnija jonus koncentrācijā 5 g uz litru (Magnesium as modifier for the determination of barium by flame atomic emission spectrometry'. Jerrow, M. et al., Analytical Proceedings, 1991, 28, 40.).
2. Induktīvi savienotas plazmas–optiskās emisijas spektrofotometrija ir pieļaujama kā liesmas atomu absorbcijas spektrometrijas alternatīva.
32.2. Šķīstošā stroncija noteikšana
396. Ar šķīstošā stroncija noteikšanas metodi veic šķīstošā stroncija ekstrahēšanu un tā noteikšanu pigmentos, kas ir sāļu un laku formā.
397. Pigmentu ekstrahē ar 0,07 M sālsskābes šķīdumu noteiktos apstākļos, un stroncija daudzumu ekstraktā kvantitatīvi nosaka, veicot atomu absorbcijas spektrometriju.
398. Šķīstošā stroncija noteikšanas metodei izmanto šādus reaģentus1:
398.1. Absolūtais etanols.
398.2. Sālsskābes šķīdums, 0,07 M.
398.3. Kālija hlorīda 8 % (m/v) šķīdums: 16 g kālija hlorīda izšķīdina 200 ml 0,07 M sālsskābes šķīdumā.
398.4. Stroncija standartšķīdums:
398.4.1. Stroncija rezerves standartšķīdums, 1000 µg/ml 0,5 M slāpekļskābes šķīdumā (SpectrosoL vai līdzvērtīgā).
398.4.2. Stroncija standartšķīdums, 100 mg/ml: ar pipeti 10,0 ml stroncija rezerves standartšķīduma (atbilstoši šā pielikuma 398.4.1.apakšpunktam) pārnes uz 100 ml mērkolbu. Uzpilda līdz atzīmei ar 0,07 M sālsskābes šķīdumu un sajauc.
399. Šķīstošā stroncija noteikšanas metodei izmanto šādu aprīkojumu:
399.1. Laboratorijas standarta aprīkojums.
399.2. Membrānfiltrs, kura poru lielums ir 0,45 mm.
399.3. Atomu absorbcijas spektrofotometrs, kas aprīkots ar doba stroncija katoda lampu.
400. Šķīstošā stroncija noteikšanu veic šādā kārtībā:
400.1. Parauga sagatavošana (šķīdumu, kas sagatavots saskaņā ar šī pielikuma 393.1.4.apakšpunktu, izmanto šķīstošā stroncija satura noteikšanai):
400.1.1. Ar pipeti pārnes 5,0 ml kālija hlorīda šķīduma (atbilstoši šā pielikuma 398.3.apakšpunktam) un alikvoto daļu (WSr ml) atšķaidītā filtrāta (atbilstoši šā pielikuma 393.1.4.apakšpunktam) uz 100 ml mērkolbu, nodrošinot koncentrāciju 2–5 mg stroncija uz mililitru (sākumam būtu piemērota 25 ml alikvotā daļa). Uzpilda līdz atzīmei ar 0,07 M sālsskābes šķīdumu un sajauc.
400.1.2. Tajā pašā dienā, veicot atomu absorbcijas spektrometriju, nosaka stroncija koncentrāciju šķīdumā (atbilstoši šā pielikuma 400.1.1.apakšpunktam).
400.2. Atomu absorbcijas spektrometrijas apstākļi:
400.2.1. Liesma: slāpekļa oksīda–acetilēna.
400.2.2. Viļņu garums: 460,7 nm.
400.2.3. Fona korekcija: nav vajadzīga.
400.2.4. Degšanas apstākļi: nabadzīga liesma; maksimālai absorbcijai – optimāls degļa garums un degšanas apstākļi.
400.3. Kalibrēšana:
400.3.1. Ar pipeti vairākās 100 ml mērkolbās iepilda attiecīgi 1,0, 2,0, 3,0, 4,0, un 5,0 ml stroncija standartšķīduma (atbilstoši šā pielikuma 398.4.2.apakšpunktam). Uz katru kolbu ar pipeti pārnes 5,0 ml kālija hlorīda šķīduma (atbilstoši šā pielikuma 398.3.apakšpunktam), uzpilda līdz atzīmei ar 0,07 M sālsskābes šķīdumu un sajauc. Šie šķīdumi satur attiecīgi 1,0, 2,0, 3,0 4,0, un 5,0 mg stroncija uz mililitru. Līdzīgi sagatavo tukšo šķīdumu bez stroncija standartšķīduma.
400.3.2. Izmēra tukšā šķīduma (atbilstoši šā pielikuma 400.3.1.apakšpunktam) absorbciju, un iegūto vērtību kalibrēšanas līknē izmanto kā stroncija nulles koncentrāciju. Izmēra katra stroncija kalibrēšanas standarta (atbilstoši šā pielikuma 400.3.1.apakšpunktam) absorbciju. Uzzīmē kalibrēšanas līkni, attiecinot absorbcijas maksimuma vērtības pret stroncija koncentrāciju.
400.4. Noteikšana. Izmēra paraugšķīduma (atbilstoši šā pielikuma 400.1.1.apakšpunktam) absorbciju. No kalibrēšanas līknes nolasa stroncija koncentrāciju, kas atbilst parauga šķīduma absorbcijas vērtībai.
401. Šķīstošā stroncija saturu (% m/m) pigmentā aprēķina pēc formulas:
% (m/m) šķīstošā stroncija = ![]() , kur
, kur
m – analizējamā parauga (atbilstoši šā pielikuma 393.1.1.apakšpunktam) masa (gramos);
c – šķīstošā stroncija koncentrācija paraugšķīdumā (atbilstoši šā pielikuma 393.1.1.apakšpunktam) (mikrogramos uz mililitru), ko nosaka pēc kalibrēšanas līknes;
V – ekstrahējošās vielas kopējais tilpums (mililitros) (atbilstoši šā pielikuma 393.1.2.apakšpunktam);
WSr – ekstrakta tilpums (mililitros) (atbilstoši šā pielikuma 393.1.1.apakšpunktam).
402. Labākā šīs metodes atkārtojamības prognoze, ja šķīstošā stroncija saturs ir 0,6 % (m/m), ir 0,09 %4.
403. Induktīvi savienotas plazmas–optiskās emisijas spektrofotometrija ir pieļaujama liesmas atomu absorbcijas spektrometrijas alternatīva.
33. Benzilspirta pierādīšana un noteikšana kosmētikas līdzekļos
33.1. Benzilspirta pierādīšana kosmētikas līdzekļos
404. Ar benzilspirta pierādīšanas metodi pierāda benzilspirtu kosmētikas līdzekļos.
405. Benzilspirtu pierāda plānslāņa hromatogrāfijā uz silikagela plāksnēm.
406. Benzilspirta pierādīšanas metodei izmanto šādus reaģentus1:
406.1. Benzilspirts.
406.2. Hloroforms.
406.3. Absolūtais etanols.
406.4. n-Pentāns.
406.5. Attīstīšanas šķīdinātājs: dietilēteris.
406.6. Benzilspirta standartšķīdums: 100 ml mērkolbā iesver 0,1 g benzilspirta, uzpilda līdz atzīmei ar etanolu un sajauc.
406.7. Stikla plānslāņa hromatogrāfijas plāksnes 100 × 200 mm vai 200 × 200 mm, kas pārklātas ar 0,25 mm silikagela 60 F254 slāni.
406.8. Attīstītājs: 10 % (m/v) 12-molibdēnfosforskābe etanolā.
407. Benzilspirta pierādīšanas metodei izmanto šādu aprīkojumu:
407.1. Plānslāņa hromatogrāfijas standarta aprīkojums.
407.2. Hromatogrāfijas tvertne, kamera ar diviem nodalījumiem, kuru kopējie izmēri ir aptuveni 80 × 230 × 240 mm.
407.3. Hromatogrāfijas papīrs: vatmaņpapīrs vai līdzvērtīgs.
407.4. Ultravioletā lampa ar viļņu garumu 254 nm.
408. Benzilspirta pierādīšanu veic šādā kārtībā:
408.1. Parauga sagatavošana. 1,0 g analizējamā produkta iesver 10 ml mērkolbā. Pievieno 3 ml hloroforma un spēcīgi krata, līdz produkts ir disperģējies. Uzpilda līdz atzīmei ar etanolu un spēcīgi krata, līdz rodas dzidrs vai gandrīz dzidrs šķīdums.
408.2. Plānslāņa hromatogrāfija:
408.2.1. Hromatogrāfijas tvertni piesātina ar n-pentānu: aizmugurējam nodalījumam piegulošās kameras sienu izklāj ar hromatogrāfijas papīru tā, lai papīra apakšējā mala ir nodalījumā. 25 ml n-pentāna pārnes uz aizmugurējo nodalījumu, lejot šo šķīdinātāju uz hromatogrāfijas papīra redzamās virsmas. Nekavējoties uzliek vāku un atstāj tvertni uz 15 minūtēm.
408.2.2. Liek 10 ml parauga šķīduma (atbilstoši šā pielikuma 408.1.apakšpunktam) un 10 ml benzilspirta standartšķīduma (atbilstoši šā pielikuma 406.6.apakšpunktam) piemērotos punktos uz plānslāņa hromatogrāfijas plāksnes (atbilstoši šā pielikuma 406.7.apakšpunktam) sākuma līnijas. Ļauj nožūt.
408.2.3. Ar pipeti tvertnes priekšējā nodalījumā iepilina 10 ml dietilētera un tūlīt tajā pašā nodalījumā ieliek plāksni (atbilstoši šā pielikuma 408.2.2.apakšpunktam). Ātri uzliek tvertnei vāku un attīsta plāksni 150 mm garumā. Izņem plāksni no hromatogrāfijas tvertnes un ļauj tai izžūt istabas temperatūrā.
408.2.4. Apskata plāksni (atbilstoši šā pielikuma 408.2.3.apakšpunktam) ultravioletā gaismā un atzīmē violeto plankumu stāvokli. Apsmidzina plāksni ar attīstītāju (atbilstoši šā pielikuma 406.8.apakšpunktam) un 15 minūtes karsē 120 °C. Benzilspirts parādās tumšzila plankuma veidā.
408.2.5. Aprēķina benzilspirta standartšķīduma Rf vērtību. Tumšzils plankums ar tādu pašu Rf vērtību liecina par benzilspirta klātbūtni. Pierādīšanas robeža: 0,1 mg benzilspirta.
33.2. Benzilspirta noteikšana kosmētikas līdzekļos
409. Ar benzilspirta noteikšanas metodi nosaka benzilspirtu kosmētikas līdzekļos.
410. Benzilspirta daudzumu izsaka masas procentos (% m/m).
411. Paraugu ekstrahē ar metanolu, un benzilspirta daudzumu ekstraktā nosaka augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC).
412. Benzilspirta noteikšanas metodei izmanto šādus reaģentus2:
412.1. Metanols.
412.2. 4-Etoksifenols.
412.3. Benzilspirts.
412.4. Kustīgā fāze: metanols/ūdens 45:55 (v/v).
412.5. Benzilspirta rezerves šķīdums: 100 ml mērkolbā precīzi iesver aptuveni 0,1 g benzilspirta. Uzpilda līdz zīmei ar metanolu un sajauc.
412.6. Iekšējā standarta rezerves šķīdums: 100 ml mērkolbā precīzi iesver aptuveni 0,1 g 4-etoksifenola. Uzpilda līdz atzīmei ar metanolu un sajauc.
412.7. Standartšķīdumi: ar pipeti uz vairākām 25 ml mērkolbām saskaņā šajā apakšpunktā norādīto tabulu pārnes benzilspirta rezerves šķīdumu (atbilstoši šā pielikuma 412.5.apakšpunktam) un iekšējā standarta rezerves škīdumu (atbilstoši šā pielikuma 412.6.apakšpunktam). Uzpilda līdz atzīmei ar metanolu un sajauc.
|
Standartšķīdums |
Benzilspirta koncentrācija |
4-etoksifenola koncentrācija |
||
|
(412.5.apakšpunkts) pievienotais tilpums (ml) |
μg/ml (*) |
(412.6.apakšpunkts) pievienotais tilpums (ml) |
μg/ml (*) |
|
|
I |
0,5 |
20 |
2,0 |
80 |
|
II |
1,0 |
40 |
2,0 |
80 |
|
III |
2,0 |
80 |
2,0 |
80 |
|
IV |
3,0 |
120 |
2,0 |
80 |
|
V |
5,0 |
200 |
2,0 |
80 |
|
(*) Šīs vērtības atbilst to standartšķīdumu koncentrācijai, kas ir gatavoti, izmantojot benzilspirta šķīdumu (412.5.apakšpunkts.) un 4-etoksifenola (atbilstoši šā pielikuma 412.6.apakšpunktam) šķīdumu, kurš attiecīgi satur tieši 0,1 % (m/v) benzilspirta un tieši 0,1 % (m/v) 4-etoksifenola. |
||||
413. Benzilspirta noteikšanas metodei izmanto šādu aprīkojumu:
413.1. Laboratorijas standarta aprīkojums.
413.2. Augstas izšķirtspējas šķidruma hromatogrāfijas iekārta ar maināma viļņu garuma ultravioleto staru detektoru un 10 ml injekcijas cilpu.
413.3. Analītiskā kolonna: 250 × 4,6 mm nerūsējoša tērauda kolonna, kas pildīta ar 5 mm Spherisorb ODS vai līdzvērtīgu.
413.4. Ūdens vanna.
413.5. Ultraskaņas vanna.
413.6. Centrifūga.
413.7. Centrifūgas mēģenes, tilpums 15 ml.
414. Benzilspirta noteikšanu veic šādā kārtībā:
414.1. Parauga sagatavošana:
414.1.1. Centrifūgas mēģenē precīzi iesver aptuveni 0,1 g (m gramu) parauga un pievieno 5 ml metanola.
414.1.2. Karsē 10 minūtes ūdens vannā 50 °C temperatūrā, pēc tam mēģeni liek ultraskaņas vannā, līdz paraugs ir pilnīgi disperģēts.
414.1.3. Atdzesē un 5 minūtes centrifugē ar 3500 apgriezieniem minūtē.
414.1.4. Centrifugātu pārnes uz 25 ml mērkolbu.
414.1.5. Atkārtoti ekstrahē paraugu ar 5 ml metanola. Ekstraktus apvieno 25 ml mērkolbā.
414.1.6. Ar pipeti pārnes 2,0 ml iekšējā standarta rezerves šķīduma (atbilstoši šā pielikuma 412.6.apakšpunktam) uz 25 ml mērkolbu. Uzpilda līdz zīmei ar metanolu un sajauc. Šo šķīdumu izmanto šā pielikuma 414.4.apakšpunktā minētajā noteikšanā.
414.2. Hromatogrāfija:
414.2.1. Uzstāda augstas izšķirtspējas šķidruma hromatogrāfijas iekārtu (atbilstoši šā pielikuma 413.2.apakšpunktam). Noregulē kustīgās fāzes (atbilstoši šā pielikuma 412.4.apakšpunktam) plūsmas ātrumu 2,0 ml minūtē.
414.2.2. Ultravioleto staru detektora viļņu garumu noregulē uz 210 nm.
414.3. Kalibrēšana:
414.3.1. No katra benzilspirta standartšķīduma (atbilstoši šā pielikuma 412.7.apakšpunktam) iesmidzina 10 ml un izmēra benzilspirta un 4-etoksifenola pīķu laukumus.
414.3.2. Katram benzilspirta standartšķīdumam (atbilstoši šā pielikuma 412.7.apakšpunktam) aprēķina benzilspirta un 4-etoksifenola pīķa laukumu attiecību. Uzzīmē kalibrēšanas līkni, šīs attiecības izmantojot kā ordinātu un attiecīgās benzilspirta koncentrācijas mg uz mililitru kā abscisu.
414.4. Noteikšana:
414.4.1. No katra benzilspirta paraugšķīduma (atbilstoši šā pielikuma 414.1.6.apakšpunktam) iesmidzina 10 ml un izmēra benzilspirta un 4-etoksifenola pīķu laukumus. Aprēķina benzilspirta un 4-etoksifenola pīķu laukumu attiecību. Šo procedūru atkārto ar parauga šķīduma 10 ml alikvotām daļām, līdz iegūst konsekventus rezultātus.
414.4.2. No kalibrēšanas līknes (atbilstoši šā pielikuma 414.3.2.apakšpunktam) nolasa benzilspirta koncentrāciju, kas atbilst benzilspirta pīķu laukuma attiecībai pret 4-etoksifenola pīķu laukumu.
415. Aprēķina benzilspirta saturu paraugā un izsaka masas procentos, izmantojot formulu:
% (m/m) benzilspirta = ![]() , kur
, kur
m – analizējamā parauga (atbilstoši šā pielikuma 414.1.1.apakšpunktam) masa (gramos);
c – benzilspirta koncentrācija paraugšķīdumā (atbilstoši šā pielikuma 414.1.6.apakšpunktam) (mikrogramos uz mililitru), ko nosaka pēc kalibrēšanas līknes.
416. No viena parauga ar 1 % (m/m) benzilspirta saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,10 %4.
34. Cirkonija pierādīšana un cirkonija, alumīnija un hlora noteikšana pretsviedru līdzekļos, kas nav aerosola formā
34.1. Cirkonija pierādīšana
417. Ar cirkonija pierādīšanas metodi pierāda cirkoniju pretsviedru kosmētikas līdzekļos, kas nav aerosola formā.
418. Cirkoniju pierāda raksturīgas sarkanvioletas nogulsnes, kas rodas ar alizarīnsarkano S stipri skābā vidē.
419. Cirkonija pierādīšanas metodei izmanto šādus reaģentus1:
419.1. Koncentrēta (d20 = 1,18 g/ml) sālsskābe;
419.2. Alizarīnsarkanā S (CI. 58005) šķīdums: 2 % (m/v) nātrija alizarīna sulfonāta ūdens šķīdums.
420. Cirkonija pierādīšanas metodei izmanto laboratorijas standarta aprīkojumu.
421. Cirkonija pierādīšanu veic šādā kārtībā:
421.1. Aptuveni 1 g parauga mēģenē pievieno 2 ml ūdens. Mēģeni noslēdz un krata.
421.2. Pievieno trīs pilienus alizarīnsarkanā S šķīduma un pēc tam 2 ml koncentrētas sālsskābes. Mēģeni noslēdz un krata.
421.3. Nostādina apmēram divas minūtes.
421.4. Sarkanviolets uzpeldējums un nogulsnes liecina par cirkonija klātbūtni.
34.2. Cirkonija noteikšana
422. Ar cirkonija noteikšanas metodi nosaka cirkoniju alumīnija cirkonija hlorīda hidroksīda kompleksos līdz maksimālajai cirkonija koncentrācijai 7,5 % (m/m) pretsviedru līdzekļos, kas nav aerosola formā.
423. Cirkoniju ekstrahē no produkta skābā vidē un nosaka, veicot liesmas atomu absorbcijas spektrometriju.
424. Cirkonija noteikšanas metodei izmanto šādus reaģentus1:
424.1. Koncentrēta (d20 = 1,18 g/ml) sālsskābe.
424.2. Sālsskābes šķīdums, 10 % (v/v): 500 ml ūdens vārglāzē, nepārtaukti maisot, pievieno 100 ml koncentrētas sālsskābes. Šo šķīdumu pārlej vienlitra mērkolbā un uzpilda līdz atzīmei ar ūdeni.
424.3. Stroncija rezerves standartšķīdums, 1000 µg/ml 0,5 M sālsskābes šķīdumā (SpectrosoL vai līdzvērtīgā).
424.4. Alumīnija hlorīda (hidrēts) [AlCl3·6H2O] reaģents: 22,6 g alumīnija hlorīda heksahidrāta izšķīdina 250 ml 10 % (v/v) sālsskābes šķīduma (atbilstoši šā pielikuma 424.2.apakšpunktam).
424.5. Amonija hlorīda reaģents: 5,0 g amonija hlorīda izšķīdina 250 ml 10 % (v/v) sālsskābes šķīduma (atbilstoši šā pielikuma 424.2.apakšpunktam).
425. Cirkonija noteikšanas metodei izmanto šādu aprīkojumu:
425.1. Laboratorijas standarta aprīkojums.
425.2. Sildītājs ar magnētisko maisītāju.
425.3. Filtrpapīrs (vatmaņpapīrs Nr.41 vai līdzvērtīgs).
425.4. Atomu absorbcijas spektrofotometrs, kas aprīkots ar doba cirkonija katoda lampu.
426. Cirkonija noteikšanu veic šādā kārtībā:
426.1. Parauga sagatavošana:
426.1.1. Precīzi iesver aptuveni 1,0 g (m gramu) homogēna produkta parauga 150 ml vārglāzē. Pievieno 40 ml ūdens un 10 ml koncentrētas sālsskābes (atbilstoši šā pielikuma 424.1.apakšpunktam).
426.1.2. Liek vārglāzi uz sildītāja ar magnētisko maisītāju. Sāk maisīt un karsē līdz viršanai. Lai novērstu strauju izžūšanu, vārglāzi pārsedz ar stiklu. Vāra piecas minūtes, noņem vārglāzi no sildītāja un atdzesē līdz istabas temperatūrai.
426.1.3. Vārglāzes saturu caur filtrpapīru izfiltrē 100 ml mērkolbā. Vārglāzi izskalo ar divām 10 ml ūdens devām un mazgājumus pēc filtrēšanas pievieno kolbas saturam. Uzpilda ar ūdeni līdz atzīmei un sajauc. Šo šķīdumu lieto arī alumīnija noteikšanai (atbilstoši šā pielikuma 34.3.apakšnodaļai).
426.1.4. Ar pipeti uz 50 ml mērkolbu pārnes 20,00 ml paraugšķīduma (atbilstoši šā pielikuma 426.1.3.apakšpunktam), 5,00 ml alumīnija hlorīda reaģenta (atbilstoši šā pielikuma 424.4.apakšpunktam) un 5,00 ml amonija hlorīda reaģenta (atbilstoši šā pielikuma 424.5.apakšpunktam). Uzpilda līdz atzīmei ar 10 % (v/v) sālsskābes šķīdumu (atbilstoši šā pielikuma 424.2.apakšpunktam) un sajauc.
426.2. Atomu absorbcijas spektrometrijas apstākļi:
426.2.1. Liesma: slāpekļa oksīda–acetilēna.
426.2.2. Viļņu garums: 360,1 nm.
426.2.3. Fona korekcija: nav vajadzīga.
426.2.4. Degšanas apstākļi: bagāta liesma; maksimālai absorbcijai – optimāls degļa garums un degšanas apstākļi.
426.3. Kalibrēšana:
426.3.1. Ar pipeti vairākās 50 ml mērkolbās iepilda attiecīgi 5,00, 10,00, 15,00, 20,00 un 25,00 ml cirkonija rezerves standartšķīduma (atbilstoši šā pielikuma 424.3.apakšpunktam). Ar pipeti katrā mērkolbā iepilda 5,00 ml alumīnija hlorīda reaģenta (atbilstoši šā pielikuma 424.4.apakšpunktam) un 5,00 ml amonija hlorīda reaģenta (atbilstoši šā pielikuma 424.5.apakšpunktam). Uzpilda līdz atzīmei ar 10 % (v/v) sālsskābes šķīdumu (atbilstoši šā pielikuma 424.2.apakšpunktam) un sajauc. Šie šķīdumi satur attiecīgi 100, 200, 300, 400 un 500 mg cirkonija uz mililitru. Līdzīgi sagatavo tukšo šķīdumu bez cirkonija standartšķīduma.
426.3.2. Izmēra tukšā šķīduma (atbilstoši šā pielikuma 426.3.1.apakšpunktam) absorbciju un iegūto vērtību kalibrēšanas līknē izmanto kā cirkonija nulles koncentrāciju. Izmēra katra cirkonija kalibrēšanas standarta (atbilstoši šā pielikuma 426.3.1.apakšpunktam) absorbciju. Uzzīmē kalibrēšanas līkni, attiecinot absorbcijas vērtības pret cirkonija koncentrāciju.
426.4. Noteikšana. Izmēra paraugšķīduma (atbilstoši šā pielikuma 426.1.4.apakšpunktam) absorbciju. No kalibrēšanas līknes nolasa cirkonija koncentrāciju, kas atbilst paraugšķīduma absorbcijas vērtībai.
427. Aprēķina cirkonija saturu paraugā masas procentos (% m/m), izmantojot formulu:
% (m/m) cirkonija = ![]() , kur
, kur
m – analizējamā parauga (atbilstoši šā pielikuma 426.1.1.apakšpunktam) masa (gramos);
c – cirkonija koncentrācija paraugšķīdumā (atbilstoši šā pielikuma 426.1.4.apakšpunktam) (mikrogramos uz mililitru), ko nosaka pēc kalibrēšanas līknes.
428. No viena parauga ar 3,00 % (m/m) cirkonija saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,10 % (m/m)4.
429. Induktīvi savienotas plazmas – optiskās emisijas spektrometrija ir pieļaujama liesmas atomu absorbcijas spektrometrijas alternatīva.
34.3. Alumīnija noteikšana
430. Ar alumīnija noteikšanas metodi nosaka alumīniju alumīnija cirkonija hlorīda hidroksīda kompleksos līdz maksimālajai alumīnija koncentrācijai 12 % (m/m) pretsviedru līdzekļos, kas nav aerosola formā.
431. Alumīniju ekstrahē no produkta skābā vidē un nosaka, veicot liesmas atomu absorbcijas spektrometriju.
432. Alumīnija noteikšanas metodei izmanto šādus reaģentus1:
432.1. Koncentrēta (d20 = 1,18 g/ml) sālsskābe.
432.2. Sālsskābes šķīdums, 1 % (v/v): 500 ml ūdens vārglāzē, nepārtaukti maisot, pievieno 10 ml koncentrētas sālsskābes. Šo šķīdumu pārlej vienlitra mērkolbā un uzpilda līdz atzīmei ar ūdeni.
432.3. Alumīnija rezerves standartšķīdums, 1000 µg/ml 0,5 M slāpekļskābes šķīdumā (SpectrosoL vai līdzvērtīgā).
432.4. Kālija hlorīda reaģents: 10,0 g kālija hlorīda izšķīdina 250 ml 1 % (v/v) sālsskābes šķīduma.
433. Alumīnija noteikšanas metodei izmanto šādu aprīkojumu:
433.1. Laboratorijas standarta aprīkojums.
433.2. Atomu absorbcijas spektrofotometrs, kas aprīkots ar doba alumīnija katoda lampu.
434. Alumīnija noteikšanu veic šādā kārtībā:
434.1. Parauga sagatavošana:
434.1.1. Alumīnija satura noteikšanai izmanto šķīdumu, kas sagatavots saskaņā ar šī pielikuma 426.1.3.apakšpunktu.
434.1.2. Ar pipeti 100 ml mērkolbā pārnes 5,00 ml paraugšķīduma un 10,00 ml kālija hlorīda reaģenta. Uzpilda līdz atzīmei ar 1 % (v/v) sālsskābes šķīdumu un sajauc.
434.2. Atomu absorbcijas spektrometrijas apstākļi:
434.2.1. Liesma: slāpekļa oksīda–acetilēna.
434.2.2. Viļņu garums: 309,3 nm.
434.2.3. Fona korekcija: nav vajadzīga.
434.2.4. Degšanas apstākļi: bagāta liesma; maksimālai absorbcijai – optimāls degļa garums un degšanas apstākļi.
434.3. Kalibrēšana:
434.3.1. 100 ml mērkolbās iepilda attiecīgi 1,00, 2,00, 3,00, 4,00 un 5,00 ml alumīnija rezerves standartšķīduma. Ar pipeti uz katru mērkolbu pārnes 10,00 ml kālija hlorīda reaģenta, uzpilda līdz atzīmei ar 1 % (v/v) sālsskābes šķīdumu un sajauc. Šie šķīdumi satur 10, 20, 30, 40 un 50 µg alumīnija uz mililitru. Līdzīgi sagatavo tukšo šķīdumu bez alumīnija standartšķīduma.
434.3.2. Izmēra tukšā šķīduma absorbciju un iegūto vērtību kalibrēšanas līknē izmanto kā alumīnija nulles koncentrāciju. Izmēra katra alumīnija kalibrēšanas standarta absorbciju. Uzzīmē kalibrēšanas līkni, attiecinot absorbcijas vērtības pret alumīnija koncentrāciju.
434.4. Noteikšana:
434.4.1. Izmēra paraugšķīduma absorbciju.
434.4.2. No kalibrēšanas līknes nolasa alumīnija koncentrāciju, kas atbilst parauga šķīduma absorbcijas vērtībai.
435. Aprēķina alumīnija saturu paraugā masas procentos pēc formulas:
% (m/m) alumīnija = ![]() , kur
, kur
m – noteikšanai ņemtā parauga masa (gramos);
c – alumīnija koncentrācija paraugšķīdumā (mikrogramos uz mililitru), ko nosaka pēc kalibrēšanas līknes.
436. No viena parauga ar 3,5 % (m/m) alumīnija saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,10 % (m/m) 4.
437. Induktīvi savienotas plazmas–optiskās emisijas spektrofotometrija ir pieļaujama liesmas atomu absorbcijas spektrometrijas alternatīva.
34.4. Hlora noteikšana
438. Ar hlora noteikšanas metodi nosaka hloru kā hlorīda jonu alumīnija cirkonija hlorīda hidroksīda kompleksos pretsviedru līdzekļos, kas nav aerosola formā.
439. Hlorīda jonus produktā nosaka, potenciometriski titrējot ar standarta sudraba nitrāta šķīdumu.
440. Hlora noteikšanas metodei izmanto šādus reaģentus1:
440.1. Koncentrēta slāpekļskābe (d20 = 1,42 g/ml).
440.2. Slāpekļskābes šķīdums, 5 % (v/v): 250 ml ūdens vārglāzē, nepārtraukti maisot, pievieno 25 ml koncentrētas slāpekļskābes. Šo šķīdumu pārnes uz 500 ml mērkolbu un uzpilda līdz atzīmei ar ūdeni.
440.3. Acetons.
440.4. Sudraba nitrāts, 0,1 M volumetrisks šķīdums (AnalaR vai līdzvērtīgs).
441. Hlora noteikšanas metodei izmanto šādu aprīkojumu:
441.1. Standarta laboratorijas aprīkojums.
441.2. Sildītājs ar magnētisko maisītāju.
441.3. Sudraba elektrods.
441.4. Kalomela standartelektrods.
441.5. pH/milivoltmetrs, kas piemērots potenciometriskai titrēšanai.
442. Hlora noteikšanu veic šādā kārtībā:
442.1. Parauga sagatavošana:
442.1.1. Precīzi iesver aptuveni 1,0 g (m gramu) homogēna produkta parauga 250 ml vārglāzē. Pievieno 80 ml ūdens un 20 ml 5 % (v/v) slāpekļskābes šķīduma;
442.1.2. Liek vārglāzi uz sildītāja ar magnētisko maisītāju. Sāk maisīt un karsē līdz viršanai. Lai novērstu strauju izžūšanu, vārglāzi pārsedz ar stiklu. Vāra piecas minūtes, noņem vārglāzi no sildītāja un atdzesē līdz istabas temperatūrai;
442.1.3. Pievieno 10 ml acetona, iegremdē elektrodus (atbilstoši šā pielikuma 441.3. un 441.4.apakšpunktam) zem šķīduma virsmas un sāk maisīt. Potenciometriski titrē ar 0,1 M sudraba nitrāta šķīdumu un uzzīmē diferenciālu līkni, lai noteiktu līdzsvara punktu (V ml).
443. Aprēķina hlora saturu paraugā masas procentos, izmantojot formulu:
% (m/m) hlora = ![]() , kur
, kur
m – noteikšanai ņemtā parauga masa (gramos);
v – 0,1 M sudraba nitrāta tilpums (mililitros), titrējot līdzsvara punktā.
444. No viena parauga ar 4 % (m/m) hlora saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,10 % (m/m) 4.
34.5. Alumīnija atomu attiecības pret cirkonija atomiem un alumīnija un cirkonija atomu attiecības pret hlora atomiem aprēķins
445. Alumīnija atomu attiecības pret cirkonija atomiem aprēķins. Al:Zr attiecību aprēķina pēc formulas:
Al:Zr attiecība = ![]()
446. Alumīnija un cirkonija atomu attiecības pret hlora atomiem aprēķins. (Al+Zr):Cl attiecību aprēķina pēc formulas:
Al:Zr attiecība = 
35. Heksamidīna, dibromheksamidīna, dibrompropamidīna un hlorheksidīna pierādīšana un noteikšana
447. Ar heksamidīna, dibromheksamidīna, dibrompropamidīna un hlorheksidīna pierādīšanas un noteikšanas metodi kosmētikas līdzekļos kvantitatīvi un kvalitatīvi nosaka heksamidīnu un tā sāļus, tai skaitā izetionātu un 4-hidroksibenzoātu, dibromheksamidīnu un tā sāļus, tai skaitā izetionātu, dibrompropamidīnu un tā sāļus, tai skaitā izetionātu, hlorheksidīna diacetātu, diglukonātu un dihidrohlorīdu.
448. Heksamidīna, dibromheksamidīna, dibrompropamidīna un hlorheksidīna koncentrāciju izsaka masas procentos (% m/m).
449. Pierādīšanu un noteikšanu veic jonu pāru apgrieztās fāzes augstas izšķirtspējas šķidruma hromatogrāfijā (HPLC), kam seko ultravioletā spektrofotometrija. Heksamidīnu, dibromheksamidīnu, dibrompropamidīnu un hlorheksidīnu pierāda izdalīšanas laiks hromatogrāfijas kolonnā.
450. Heksamidīna, dibromheksamidīna, dibrompropamidīna un hlorheksidīna pierādīšanas un noteikšanas metodei izmanto šādus reaģentus1, 2:
450.1. Metanols.
450.2. 1-Heptānsulfoskābes nātrija sāls monohidrāts.
450.3. Ledus etiķskābe (d20 = 1,05 g/ml).
450.4. Nātrija hlorīds.
450.5. Kustīgās fāzes:
450.5.1. Šķīdinātājs I: 0,005 M 1-heptānsulfoskābes nātrija sāls monohidrāta (atbilstoši šā pielikuma 450.2.apakšpunktam) šķīdums metanolā (atbilstoši šā pielikuma 450.1.apakšpunktam), kas līdz pH 3,5 koriģēts ar ledus etiķskābi (atbilstoši šā pielikuma 450.3.apakšpunktam).
450.5.2. Šķīdinātājs II: 0,005 M 1-heptānsulfoskābes nātrija sāls monohidrāta (450.2.apakšpunkts) šķīdums ūdenī, kas līdz pH 3,5 koriģēts ar ledus etiķskābi (450.3.apakšpunkts).
450.5.3. Ja jāuzlabo pīķu forma, kustīgās fāzes var modificēt un gatavot šādi:
450.5.3.1. Šķīdinātājs I: izšķīdina 5,84 g nātrija hlorīda un 1,1013 g
1-heptānsulfoskābes nātrija sāls monohidrāta 100 ml ūdens. Pievieno 900 ml metanola un ar ledus etiķskābi (atbilstoši šā pielikuma 450.3.apakšpunktam) koriģē līdz pH 3,5.
450.5.3.2. Šķīdinātājs II: 5,84 g nātrija hlorīda un 1,1013 g 1-heptānsulfoskābes nātrija sāls monohidrāta izšķīdina vienā litrā ūdens un koriģē ar ledus etiķskābi līdz pH 3,5.
450.6. Heksamidīna diizetionāts [C20H26N4O2·2C2H6O4S].
450.7. Dibromheksamidīna diizetionāts [C20H24Br2N4O2·2C2H6O4S].
450.8. Dibrompropamidīna diizetionāts [C17H18Br2N4O2·2C2H6O4S].
450.9. Hlorheksidīna diacetāts [C22H30Cl2N10·2C2H4O2].
450.10. Standartšķīdumi: sagatavo attiecīgi visu četru konservantu (atbilstoši šā pielikuma 450.6., 450.7., 450.8. un 450.9.apakšpunktam) 0,05 % (m/v) šķīdumus šķīdinātājā I (atbilstoši šā pielikuma 450.5.1.apakšpunktam).
450.11. 3,4,4’-trihlorkarbanilīds (triklokarbāns).
450.12. 4,4’-dihlor-3-(trifluormetil)karbanilīds (halokarbāns).
451. Heksamidīna, dibromheksamidīna, dibrompropamidīna un hlorheksidīna pierādīšanas un noteikšanas metodei izmanto šādu aprīkojumu:
451.1. Standarta laboratorijas aprīkojums.
451.2. Augstas izšķirtspējas šķidruma hromatogrāfs ar maināma viļņu garuma ultravioleto staru detektoru.
451.3. Hromatogrāfijas kolonna: nerūsējoša tērauda, 30 cm gara, ar 4 mm iekšējo diametru, pildīta ar 10 μm μ–Bondapack C18 vai līdzvērtīgu.
451.4. Ultraskaņas vanna.
452. Heksamidīna, dibromheksamidīna, dibrompropamidīna un hlorheksidīna kvalitatīvu noteikšanu veic šādā kārtībā:
452.1. Parauga sagatavošana:
452.1.1. Precīzi iesver aptuveni 0,5 g parauga 10 ml mērkolbā un uzpilda līdz atzīmei ar šķīdinātāju I (atbilstoši šā pielikuma 450.5.1.apakšpunktam).
452.1.2. Kolbu uz 10 minūtēm ieliek ultraskaņas vannā.
452.1.3. Šķīdumu centrifugē vai izfiltrē.
452.1.4. Filtrātu vai centrifugātu savāc hromatogrāfijai.
452.2. Hromatogrāfija:
452.2.1. Mobilās fāzes gradients.
|
Laiks (min.) |
Šķīdinātājs I (% v/v) (atbilstoši šā pielikuma 450.5.1.apakšpunktam) |
Šķīdinātājs II (% v/v) (atbilstoši šā pielikuma 450.5.2.apakšpunktam) |
|
0 |
50 |
50 |
|
15 |
65 |
35 |
|
30 |
65 |
35 |
|
45 |
50 |
50 |
452.2.2. Kustīgās fāzes (atbilstoši šā pielikuma 452.2.1.apakšpunktam) plūsmas ātrumu noregulē 1,5 ml/min un kolonnas temperatūru 35 °C.
452.2.3. Detektora viļņu garumu noregulē 264 nm.
452.2.4. No katra standartšķīduma (atbilstoši šā pielikuma 450.10.apakšpunktam) iesmidzina 10 μl un reģistrē to hromatogrammas.
452.2.5. Iesmidzina 10 μl paraugšķīduma (452.1.apakšpunktam) un reģistrē tā hromatogrammu.
452.3. Salīdzinot šā pielikuma 452.2.5.apakšpunktā minētā(-o) reģistrētā(-o) pīķa(-u) izdalīšanas laiku(-s) ar to(-iem) laiku(-iem), kas iegūts(-i) attiecībā uz standartšķīdumiem (atbilstoši šā pielikuma 452.2.4.apakšpunktam), pierāda heksamidīna, dibromheksamidīna, dibrompropamidīna vai hlorheksidīna klātbūtni.
453. Heksamidīna, dibromheksamidīna, dibrompropamidīna un hlorheksidīna kvantitatīvu noteikšanu veic šādā kārtībā:
453.1. Gatavojot standarta šķīdumu, izmanto vienu no konservantiem (atbilstoši šā pielikuma 450.6., 450.7., 450.8. vai 450.9.apakšpunktam), kura nav paraugā iekšējā standarta veidā. Ja tas nav iespējams, var izmantot triklokarbānu vai halokarbānu:
453.1.1. Šī pielikuma 452.3.apakšpunktā pierādītā konservanta 0,05 % (m/v) rezerves šķīdums šķīdinātājā I (atbilstoši šā pielikuma 450.5.1.apakšpunktam).
453.1.2. 0,05 % (m/v) rezerves šķīdums konservanta, kas izvēlēts kā iekšējais standarts, šķīdinātājā I (atbilstoši šā pielikuma 450.5.1.apakšpunktam).
453.1.3. Katram pierādītajam konservantam sagatavo četrus standartšķīdumus, pārnesot uz vairākām 10 ml mērkolbām attiecīgo konservantu (atbilstoši šā pielikuma 453.1.1.apakšpunktam) rezerves šķīdumus un attiecīgus daudzumus iekšējā standarta rezerves šķīduma (atbilstoši šā pielikuma 453.1.2.apakšpunktam) saskaņā ar šajā punktā norādīto tabulu. Katru kolbu uzpilda līdz atzīmei ar šķīdinātāju I (atbilstoši šā pielikuma 450.5.1.apakšpunktam) un sajauc.
|
Standartšķīdums |
Iekšējā standarta rezerves šķīdums |
Pierādītā konservanta rezerves šķīdums |
|
|
(atbilstoši šā pielikuma 453.1.2.apakšpunktam) pievienotie ml |
(atbilstoši šā pielikuma 453.1.1.apakšpunktam) pievienotie ml |
μg/ml (*) |
|
|
I |
1,0 |
0,5 |
25 |
|
II |
1,0 |
1,0 |
50 |
|
III |
1,0 |
1,5 |
75 |
|
IV |
1,0 |
2,0 |
100 |
|
(*) Šīs vērtības atbilst pierādīto konservantu koncentrācijai standartšķīdumos, kas gatavoti, izmantojot rezerves šķīdumu, kas satur tieši 0,05 % pierādītā konservanta. |
|||
453.2. Parauga sagatavošana:
453.2.1. Precīzi iesver aptuveni 0,5 g (p gramus) parauga 10 ml mērkolbā, pievieno 1,0 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 453.1.2.apakšpunktam) un 6 ml šķīdinātāja I (atbilstoši šā pielikuma 450.5.1.apakšpunktam) un sajauc.
453.2.2. Kolbu uz 10 minūtēm ieliek ultraskaņas vannā. Atdzesē. Uzpilda līdz atzīmei ar šķīdinātāju I un sajauc. Centrifugē vai filtrē caur kroku filtrpapīru. Savāc attiecīgi centrifugātu vai filtrātu hromatogrāfijai.
453.3. Hromatogrāfija:
453.3.1. Noregulē mobilās fāzes gradientu, plūsmas ātrumu, kolonnas temperatūru un HPLC iekārtas detektora viļņu garumu atbilstīgi nosacījumiem pierādīšanas stadijā (atbilstoši šā pielikuma 452.2.1., 452.2.2. un 452.2.3.apakšpunktam).
453.3.2. Iesmidzina 10 ml paraugšķīduma (atbilstoši šā pielikuma 453.2.2.apakšpunktam) un izmēra pīķu laukumus. Šo procedūru atkārto ar parauga šķīduma 10 ml alikvotām daļām, līdz iegūst konsekventus rezultātus. Aprēķina tā pīķa laukuma, kas radies no analizējamā savienojuma, attiecību pret tā pīķa laukumu, ko rada iekšējais standarts.
453.4. Kalibrēšana:
453.4.1. Iesmidzina 10 ml no katra standartšķīduma (atbilstoši šā pielikuma 453.1.3.apakšpunktam) un izmēra pīķu laukumus.
453.4.2. Atbilstīgi katram standartšķīdumam (atbilstoši šā pielikuma 453.1.3.apakšpunktam) aprēķina heksamidīna, dibromheksamidīna, dibrompropamidīna vai hlorheksidīna pīķu laukuma attiecību pret iekšējā standarta pīķu laukumu. Uzzīmē kalibrēšanas līkni, šīs attiecības izmantojot par ordinātu un attiecīgās kvalitatīvi noteiktā konservanta koncentrācijas standartšķīdumos mikrogramos uz mililitru – par abscisu.
453.4.3. No kalibrēšanas līknes (atbilstoši šā pielikuma 453.4.2.apakšpunktam) nolasa pierādītā konservanta koncentrāciju, kas atbilst pīķu laukumu attiecībai, kas aprēķināta saskaņā ar šā pielikuma 453.3.2.apakšpunktu.
454. Aprēķina heksamidīna, dibromheksamidīna, dibrompropamidīna vai hlorheksidīna saturu paraugā masas procentos, izmantojot formulu:
% (m/m) = ![]() , kur
, kur
p – parauga (atbilstoši šā pielikuma 453.2.1.apakšpunktam) masa (gramos);
c – konservanta koncentrācija paraugšķīdumā (mikrogramos uz mililitru), ko nosaka pēc kalibrēšanas līknes;
MW1 – konservanta pamatformas molekulārais svars;
MW2 – attiecīgā sāls molekulārais svars (atbilstoši šā pielikuma 456.punktam).
455. No viena parauga ar 0,1 % (m/m) heksamidīna, dibromheksamidīna, dibrompropamidīna vai hlorheksidīna saturu divās paralēlās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,005 %4.
456. Molekulāro svaru tabula
| Heksamidīns | C20H26N4O2 |
354,45 |
| Heksamidīna diizetionāts | C20H26N4O2·2C2H6O4S |
606,72 |
| Heksamidīna di-p-hidroksibenzoāts | C20H26N4O2·2C7H6O3 |
630,71 |
| Dibromheksamidīns | C20H24Br2N4O2 |
512,24 |
| Dibromheksamidīna diizetionāts | C20H24Br2N4O2·2C2H6O4S |
764,51 |
| Dibrompropamidīns | C17H18Br2N4O2 |
470,18 |
| Dibrompropamidīna diizetionāts | C17H18Br2N4O2·2C2H6O4S |
722,43 |
| Hlorheksidīns | C22H30Cl2N10 |
505,45 |
|
Hlorheksidīna diacetāts |
C22H30Cl2N10·2C2H4O2 |
625,56 |
| Hlorheksidīna diglukonāts | C22H30Cl2N10·2C6H12O7 |
897,76 |
| Hlorheksidīna dihidrohlorīds | C22H30Cl2N10·2HCl |
578,37 |
36. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes, salicilskābes un propionskābes kvalitatīva un kvantitatīva noteikšana kosmētikas līdzekļos
457. Ar benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes, salicilskābes un propionskābes kvalitatīvas un kvantitatīvas noteikšanas metodi kvalitatīvi un kvantitatīvi nosaka benzoskābi, 4-hidroksibenzoskābi, sorbīnskābi, salicilskābi un propionskābi kosmētikas līdzekļos. Atsevišķas procedūras ir noteiktas šo konservantu kvalitatīvai noteikšanai, propionskābes kvantitatīvai noteikšanai, kā arī 4-hidroksibenzoskābes, salicilskābes, sorbīnskābes un benzoskābes kvantitatīvai noteikšanai.
458. Kvantitatīvi noteiktos benzoskābes, 4-hidroksibenzoskābes, salicilskābes un propionskābes daudzumus izsaka brīvo skābju masas procentos.
36.1. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes, salicilskābes un propionskābes kvalitatīva noteikšana
459. Pēc konservantu ekstrakcijas ar skābi/bāzi ekstraktu analizē plānslāņa hromatogrāfijā, derivatizējot uz plāksnītes. Atkarībā no rezultātiem kvantitatīvas noteikšanas rezultātu apstiprina augstas izšķirtspējas šķidruma hromatogrāfijā vai – propionskābei – gāzu hromatogrāfijā.
460. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes, salicilskābes un propionskābes kvalitatīvas noteikšanas metodei izmanto šādus reaģentus1, 3:
460.1. Acetons.
460.2. Dietilēteris.
460.3. Acetonitrils.
460.4. Toluols.
460.5. n-heksāns.
460.6. Šķidrs parafīns.
460.7. 4 M sālsskābe.
460.8. 4 M kālija hidroksīda šķīdums.
460.9. Kalcija hlorīds, CaCl2·2H2O.
460.10. Litija karbonāts, Li2CO3.
460.11. 2-brom-2'-acetonaftons.
460.12. 4-hidroksibenzoskābe.
460.13. Salicilskābe.
460.14. Benzoskābe.
460.15. Sorbīnskābe.
460.16. Propionskābe.
460.17. Standartšķīdumi: sagatavo visu piecu konservantu (atbilstoši šā pielikuma 460.12., 460.13., 460.14., 460.15. un 460.16.apakšpunktam) 0,1 % (m/v) šķīdumus dietilēterī.
460.18. Derivatizācijas reaģents: 0,5 % (m/v) 2-brom-2'-acetonaftona šķīdums acetonitrilā (50 mg/10 ml). Šo šķīdumu gatavo katru nedēļu un glabā ledusskapī.
460.19. Katalizatora šķīdums: 0,3 % (m/v) litija karbonāta šķīdums ūdenī (300 mg/100 ml). Šo šķīdumu gatavo tieši pirms izmantošanas.
460.20. Attīstošais šķīdinātājs: toluols/acetons 20:0,5 (v/v).
460.21. Šķidrs parafīns/n-heksāns 1:2 (v/v).
461. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes, salicilskābes un propionskābes kvalitatīvas noteikšanas metodei izmanto parasto laboratorijas aprīkojumu un šādu iekārtu:
461.1. Ūdens vanna, kurā var uzturēt 60 °C temperatūru.
461.2. Attīstīšanas kamera.
461.3. Ultravioletās gaismas avots, 254 un 366 nm.
461.4. Plānslāņu plates, Kieselgel 60, bez fluorescenta indikatora, 20 × 20 cm, slāņa biezums 0,25 mm, koncentrācijas zona 2,5 × 20 cm (Merck 11845 vai līdzvērtīgas).
461.5. Mikrošļirce, 10 µl.
461.6. Mikrošļirce, 25 µl.
461.7. Žāvēšanas skapis, kurā var uzturēt 105 °C temperatūru.
461.8. Stikla mēģenes, 50 ml, ar skrūvējamu vāciņu.
461.9. Filtrpapīrs ar 90 mm diametru, Schleicher & Schull, Weissband No 5892 vai līdzvērtīgs.
461.10. Universālais pH indikatorpapīrs, pH 1–11.
461.11. Stikla parauga mēģenes, 5 ml.
461.12. Rotācijas ietvaicētājs (Rotavapor vai līdzvērtīgs).
461.13. Elektriskā plītiņa.
462. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes, salicilskābes un propionskābes kvalitatīvu noteikšanu veic šādā kārtībā:
462.1. Parauga gatavošana:
462.1.1. Iesver aptuveni 1 g parauga 50 ml stikla mēģenē ar skrūvējamu vāciņu. Pievieno četrus pilienus 4 M sālsskābes un 40 ml acetona. Stipri bāziskiem kosmētikas līdzekļiem (piemēram, tualetes ziepēm) būtu jāpievieno 20 pilieni 4 M sālsskābes. Ar indikatorpapīru pārbauda, vai pH ir aptuveni 2. Noslēdz mēģeni un vienu minūti spēcīgi krata.
462.1.2. Ja konservantu ekstrakcija acetona fāzē jāpaātrina, maisījumu uzmanīgi uzsilda līdz apmēram 60 °C, lai iegūtu šķidru fāzi.
462.1.3. Atdzesē šķīdumu līdz istabas temperatūrai un izfiltrē caur filtrpapīru koniskā kolbā.
462.1.4. Pārnes 20 ml filtrāta uz 200 ml konisko kolbu, pievieno 20 ml ūdens un sajauc. Ar 4 M kālija hidroksīdu noregulē maisījuma pH aptuveni uz 10, mērot pH ar indikatorpapīru.
462.1.5. Pievieno 1 g kalcija hlorīda un spēcīgi sakrata. Izfiltrē caur filtrpapīru 250 ml dalāmajā piltuvē, kur ir 75 ml dietilētera, un spēcīgi krata vienu minūti. Ļauj, lai atdalās ūdens fāze, un nosūc to 250 ml koniskajā kolbā. Izlej ētera slāni. Izmantojot indikatorpapīru, noregulē ūdens šķīduma pH aptuveni uz divi ar 4 M sālsskābes šķīdumu. Pievieno 10 ml dietilētera, noslēdz kolbu un spēcīgi krata vienu minūti. Ļauj, lai atdalās ētera slānis, un pārnes to uz rotācijas ietvaicētāju. Izlej ūdens slāni.
462.1.6. Ietvaicē ētera slāni gandrīz sausu un atlikumu vēlreiz izšķīdina 1 ml dietilētera. Pārnes šķīdumu uz parauga mēģeni.
462.2. Plānslāņa hromatogrāfija:
462.2.1. Katram standartam un paraugam, kuram paredzēts uzņemt hromatogrammu, ar šļirci vienādos attālumos uz plānslāņa hromatogrāfijasplates (atbilstoši šā pielikuma 462.4.apakšpunktam) starta līnijas koncentrācijas zonā uznes 3 µl litija karbonāta šķīduma (atbilstoši šā pielikuma 460.19.apakšpunktam) un žāvē auksta gaisa plūsmā.
462.2.2. Pārnes plānslāņa hromatogrāfijasplati uz plītiņu, kas sakarsēta līdz 40 °C, lai plankumus saglabātu pēc iespējas mazus. Ar mikrošļirci uznes 10 µl katra standartšķīduma (atbilstoši šā pielikuma 460.17.apakšpunktam) un parauga šķīduma (atbilstoši šā pielikuma 462.1.apakšpunktam) uz plates starta līnijas tieši tajos punktos, kur uzlikts litija karbonāta šķīdums.
462.2.3. Beidzot uznes uz plates apmēram 15 µl derivatizācijas reaģenta (atbilstoši šā pielikuma 460.18.apakšpunktam) (2-brom-2'-acetonaftona šķīduma) tieši tajos punktos, kur uzlikti standarta/parauga šķīdumi un litija karbonāta šķīdums.
462.2.4. Plānslāņa hromatogrāfijasplati 45 minūtes karsē žāvēšanas skapī 80 °C. Kad plate atdzesēta, kamerā, kas 15 minūtes stabilizēta (bez filtrpapīra izklājuma), to attīsta ar šī pielikuma 460.20.apakšpunktā norādīto attīstīšanas šķīdinātāju (toluols/acetons), līdz šķīdinātāja fronte pavirzās par 15 cm (var būt vajadzīgas apmēram 80 minūtes).
462.2.5. Atdzesē plati auksta gaisa plūsmā un apskata iegūtos plankumus ultravioletā gaismā. Lai uzlabotu vājo plankumu fluorescenci, plānslāņa hromatogrāfijas plati var iegremdēt šķidrā parafīnā/n-heksānā (atbilstoši šā pielikuma 460.21.apakšpunktam).
463. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes, salicilskābes un propionskābes kvalitatīvas noteikšanas aprēķinu veic šādā kārtībā:
463.1. Aprēķina katra plankuma Rf vērtību.
463.2. Salīdzina parauga un standarta šķīdumu Rf un izmaiņas ultravioletajos staros.
463.3. Izdara iepriekšēju secinājumu par konservantu klātbūtni un identitāti. Veic augstas izšķirtspējas plānslāņa hromatogrāfiju saskaņā ar šā pielikuma 36.2.apakšnodaļu, vai – ja izrādās, ka paraugā ir propionskābe – gāzu hromatogrāfiju saskaņā ar šā pielikuma 36.3.apakšnodaļu. Salīdzina izdalīšanas laikus ar standarta šķīdumu izdalīšanas laikiem.
463.4. Galīgi kvalitatīvi nosakot konservantus paraugā, ņem vērā plānslāņa hromatogrāfijas un augstas izšķirtspējas plānslāņa hromatogrāfijas vai gāzu hromatogrāfijas rezultātus.
36.2. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes un salicilskābes kvantitatīva noteikšana*
464. Pēc skābināšanas paraugu ekstrahē ar etanola un ūdens maisījumu. Pēc filtrēšanas konservantus kvantitatīvi nosaka augstas izšķirtspējas šķidruma hromatogrāfijā.
465. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes un salicilskābes kvantitatīvas noteikšanas metodei izmanto šādus reaģentus1, 2, 3:
465.1. Absolūtais spirts.
465.2. 4-hidroksibenzoskābe.
465.3. Salicilskābe.
465.4. Benzoskābe.
465.5. Sorbīnskābe.
465.6. Nātrija acetāts, (CH3COONa·3H2O).
465.7. Etiķskābe, ![]() = 1,05 g/ml.
= 1,05 g/ml.
465.8. Acetonitrils.
465.9. 2 M sērskābe.
465.10. 0,2 M kālija hidroksīda šķīdums.
465.11. 2-metoksibenzoskābe.
465.12. Etanola/ūdens maisījums: sajauc deviņas tilpuma vienības etanola un vienu tilpuma vienību ūdens (2:1).
465.13. Iekšējā standarta šķīdums: sagatavo šķīdumu, kas satur apmēram 1 g 2-metoksibenzoskābes 500 mililitros etanola/ūdens maisījuma (atbilstoši šā pielikuma 465.12.apakšpunktam).
465.14. Kustīgā fāze augstas izšķirtspējas plānslāņa hromatogrāfijai:
465.14.1. Acetāta buferšķīdums: 1 l ūdens pievieno 6,35 g nātrija acetāta un 20,0 ml etiķskābes un sajauc.
465.14.2. Sagatavo kustīgo fāzi, sajaucot deviņas tilpuma vienības acetāta buferšķīduma un vienu tilpuma vienību acetonitrila.
465.15. Konservanta standartšķīdums: precīzi iesver aptuveni 0,05 g 4-hidroksibenzoskābes, 0,2 g salicilskābes, 0,2 g benzoskābes un 0,05 g sorbīnskābes 50 ml mērkolbā un uzpilda līdz zīmei ar etanola/ūdens maisījumu (atbilstoši šā pielikuma 465.12.apakšpunktam). Šo šķīdumu glabā ledusskapī. Šķīdums ir stabils vienu nedēļu.
465.16. Konservantu standartšķīdumi: attiecīgi 8,00, 4,00, 2,00, 1,00 un 0,50 ml standartšķīduma (atbilstoši šā pielikuma 465.15.apakšpunktam) pārnes uz 20 ml mērkolbām. Katrā kolbā pievieno 10,00 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 465.13.apakšpunktam) un 0,5 ml 2 M sērskābes. Uzpilda līdz zīmei ar etanola/ūdens maisījumu (atbilstoši šā pielikuma 465.12.apakšpunktam). Šie šķīdumi jāgatavo tieši pirms izmantošanas.
466. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes un salicilskābes kvantitatīvas noteikšanas metodei izmanto parasto laboratorijas aprīkojumu un šādu iekārtu:
466.1. ūdens vanna, kurā temperatūra noregulēta uz 60 °C.
466.2. augstas izšķirtspējas šķidruma hromatogrāfs ar maināma viļņu garuma ultravioleto staru detektoru un 10 µl ievadīšanas cilpu.
466.3. hromatogrāfijas kolonna – nerūsējoša tērauda, 12,5–25 cm gara, ar 4,6 mm iekšējo diametru un Nucleosil 5C18 vai līdzvērtīgu pildījumu.
466.4. filtrpapīrs ar 90 mm diametru, Schleicher & Schull, Weissband No 5892 vai līdzvērtīgs.
466.5. stikla mēģenes, 50 ml, ar skrūvējamu vāciņu.
466.6. stikla parauga mēģenes, 5 ml.
466.7. vārķermeņi, 2–4 mm, karborunda vai līdzvērtīgi.
467. Benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes un salicilskābes kvantitatīvu noteikšanu veic šādā kārtībā:
467.1. Parauga gatavošana:
467.1.1. Parauga gatavošana, nepievienojot iekšējo standartu, notiek šādi: iesver 1 g parauga 50 ml stikla mēģenē ar skrūvējamu vāciņu. Ar pipeti mēģenē iepilina 1,00 ml 2 M sērskābes un 40,0 ml etanola/ūdens maisījuma (atbilstoši šā pielikuma 465.12.apakšpunktam). Pievieno aptuveni 1 g vārķermeņu (atbilstoši šā pielikuma 466.7.apakšpunktam), noslēdz mēģeni un spēcīgi krata vismaz vienu minūti, līdz rodas vienmērīga suspensija. Lai paātrinātu konservantu ekstrakciju etanola fāzē, liek mēģeni tieši uz piecām minūtēm ūdens vannā, kur uztur 60 °C. Tūlīt atdzesē mēģeni auksta ūdens strūklā un vienu stundu tur ekstraktu 5 °C. Izfiltrē ekstraktu caur filtrpapīru. Pārnes aptuveni 2 ml ekstrakta uz parauga mēģeni. Tur ekstraktu 5 °C un 24 stundās veic kvantitatīvu noteikšanu augstas izšķirtspējas plānslāņa hromatogrāfijā.
467.1.2. Parauga gatavošana, pievienojot iekšējo standartu, notiek šādi: ar precizitāti līdz trešajai zīmei aiz komata iesver 1±0,1 g (a grami) parauga 50 ml stikla mēģenē ar skrūvējamu vāciņu. Ar pipeti mēģenē iepilina 1,00 ml 2 M sērskābes un 30,0 ml etanola/ūdens maisījuma (atbilstoši šā pielikuma 465.12.apakšpunktam). Pievieno aptuveni 1 g vārķermeņu (atbilstoši šā pielikuma 466.7.apakšpunktam) un 10,00 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 465.13.apakšpunktam). Noslēdz mēģeni un spēcīgi krata vismaz vienu minūti, līdz rodas vienmērīga suspensija. Lai paātrinātu konservantu ekstrakciju etanola fāzē, liek mēģeni tieši uz piecām minūtēm ūdens vannā, kur uztur 60 °C. Tūlīt atdzesē mēģeni auksta ūdens strūklā, un vienu stundu tur ekstraktu 5 °C. Izfiltrē ekstraktu caur filtrpapīru. Pārnes aptuveni 2 ml filtrāta uz parauga mēģeni. Tur filtrātu 5 °C un 24 stundās veic kvantitatīvo noteikšanu augstas izšķirtspējas plānslāņa hromatogrāfijā.
467.2. Augstas izšķirtspējas šķidruma hromatogrāfija:
467.2.1. Hromatogrāfijas apstākļi:
467.2.1.1. Kustīgā fāze: acetonitrila/acetāta buferšķīdums (atbilstoši šā pielikuma 465.15.apakšpunktam).
467.2.1.2. Noregulē kustīgās fāzes plūsmas ātrumu cauri kolonnai uz 2,0±0,5 ml/minūtē.
467.2.1.3. Detektora viļņu garumu noregulē uz 240 nm.
467.2.2. Kalibrēšana:
467.2.2.1. Ievada katra konservanta standartšķīduma (atbilstoši šā pielikuma 465.16.apakšpunktam) 10 µl devu šķidruma hromatogrāfā.
467.2.2.2. Pēc iegūtajām hromatogrammām nosaka konservantu standartšķīdumu pīķu augstumu attiecības pret iekšējā standarta pīķa augstumu.
467.2.2.3. Konstruē katra konservanta līkni, attiecinot šīs attiecības pret standartšķīdumu koncentrācijām.
467.2.2.4. Nosaka, vai, kalibrējot ar standartšķīdumiem, iegūst lineāru sakarību.
467.2.3. Kvantitatīva noteikšana:
467.2.3.1. Ievada 10 µl parauga ekstrakta (atbilstoši šā pielikuma 467.1.1.apakšpunktam) šķidruma hromatogrāfā un uzņem hromatogrammu.
467.2.3.2. Ievada 10 µl viena konservanta standartšķīduma (atbilstoši šā pielikuma 465.16.apakšpunktam) un uzņem hromatogrammu.
467.2.3.3. Salīdzina iegūtās hromatogrammas. Ja parauga ekstrakta (atbilstoši šā pielikuma 467.1.1.apakšpunktam) hromatogrammā nav neviena pīķa ar aptuveni tādu pašu izdalīšanas laiku, kāds ir 2-metoksibenzoskābei (ieteicams iekšējais standarts), šķidruma hromatogrāfā ievada 10 µl parauga ekstrakta, kam pievienots iekšējais standarts (atbilstoši šā pielikuma 467.1.2.apakšpunktam), un uzņem hromatogrammu. Ja parauga ekstrakta (atbilstoši šā pielikuma 467.1.1.apakšpunktam) hromatogrammā ir traucējošas vielas pīķis, kura izdalīšanas laiks ir aptuveni vienāds ar 2-metoksibenzoskābes pīķa izdalīšanas laiku, būtu jāizvēlas cits iekšējais standarts (ja kāda analizējamā konservanta nav parauga hromatogrammā, šo konservantu var izmantot kā alternatīvu iekšējo standartu).
467.2.3.4. Nosaka, vai iegūtās standartšķīduma un parauga šķīduma hromatogrammas atbilst šādām prasībām:
467.2.3.4.1. Minimālais divu pīķu atdalījums ir vismaz 0,90 (pīķu atdalījums definēts šī pielikuma 8.attēlā).
8.attēls
Pīķu atdalījums
Ja nav panākts vajadzīgais atdalījums, izmanto efektīvāku kolonnu vai regulē kustīgās fāzes sastāvu, līdz panāk atbilstību prasībām.
467.2.3.4.2. Visu iegūto pīķu asimetrijas faktors As ir 0,9–1,5 (pīķu asimetrijas faktors definēts šī pielikuma 9.attēlā). Lai uzņemtu hromatogrammu asimetrijas faktora noteikšanai, ieteicamais pašrakstītāja ātrums ir vismaz 2 cm minūtē.
9.attēls
Pīķu asimetrijas faktors
467.2.3.4.3. Iegūst stabilu bāzes līniju.
468. Skābo konservantu koncentrāciju parauga šķīdumā aprēķina pēc konservantu pīķu augstumu attiecības pret 2-metoksibenzoskābes (iekšējā standarta) pīķa augstumu un kalibrēšanas līknes. Aprēķina benzoskābes, 4-hidroksibenzoskābes, sorbīnskābes vai salicilskābes saturu paraugā masas procentos (xi) pēc formulas:
xi % (m/m) = (100 × 20 × b)/(106 × a) = (b)/(500 × a), kur
a – analizējamā daudzuma (atbilstoši šā pielikuma 467.1.2.apakšpunktam) masa (gramos);
b – konservanta koncentrācija (µg/ml) parauga ekstraktā (atbilstoši šā pielikuma 467.1.2.apakšpunktam), ko nolasa no kalibrēšanas līknes.
469. Atkārtojamība4:
469.1. No viena parauga ar 0,40 % 4-hidroksibenzoskābes saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,035 % vērtību.
469.2. No viena parauga ar aptuveni 0,50 % benzoskābes saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,050 % vērtību.
469.3. No viena parauga ar aptuveni 0,50 % salicilskābes saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,045 % vērtību.
469.4. No viena parauga ar aptuveni 0,60 % sorbīnskābes saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,035 % vērtību.
*Piezīmes.
1. Metodes rupjas pārbaudes rezultāti liecina, ka sērskābes daudzums, ko pievieno, lai ekstrahētu skābes no parauga, ir kritisks, un parauga daudzuma robežas būtu jāsaglabā atbilstīgi priekšrakstiem.
2. Ja vēlams, var izmantot piemērotu aizsargkolonnu.
36.3. Propionskābes kvantitatīva noteikšana
470. Ar propionskābes kvantitatīvas noteikšanas metodi veic propionskābes kvantitatīvu noteikšanu kosmētikas līdzekļos, ja tās koncentrācija nepārsniedz 2 %.
471. Propionskābes koncentrāciju izsaka kosmētikas līdzekļa masas procentos (% m/m).
472. Kad propionskābe ir ekstrahēta no kosmētikas līdzekļa, to kvantitatīvi nosaka gāzu hromatogrāfijā, kā iekšējo standartu izmantojot 2-metilpropionskābi.
473. Propionskābes kvantitatīvas noteikšanas metodei izmanto šādus reaģentus3:
473.1. Etanols, 96 % (v/v).
473.2. Propionskābe.
473.3. 2 -metilpropionskābe.
473.4. Ortofosforskābe, 10 % (m/v).
473.5. Propionskābes šķīdums: precīzi iesver aptuveni 1,00 g (p grami) propionskābes 50 ml mērkolbā un uzpilda līdz zīmei ar 96 % etanolu.
473.6. Iekšējā standarta šķīdums: precīzi iesver aptuveni 1,00 g (e grami) 2-metilpropionskābes 50 ml mērkolbā un uzpilda līdz zīmei ar 96 % etanolu.
474. Propionskābes kvantitatīvas noteikšanas metodei izmanto šādu iekārtu:
474.1. Parastais laboratoriju aprīkojums.
474.2. Gāzu hromatogrāfs ar liesmas jonizācijas detektoru.
474.3. Stikla mēģene (20 × 150 mm) ar skrūvējamu vāciņu.
474.4. Ūdens vanna, kas noregulēta uz 60 °C.
474.5. 10 ml stikla šļirce ar membrānfiltru (poru diametrs 0,45 µm).
475. Propionskābes kvantitatīvu noteikšanu veic šādā kārtībā:
475.1. Parauga gatavošana:
475.1.1. Parauga gatavošana bez iekšējā standarta:
475.1.1.1. Stikla mēģenē iesver aptuveni 1 g parauga.
475.1.1.2. Pievieno 0,5 ml fosforskābes un 9,5 ml etanola.
475.1.1.3. Noslēdz mēģeni un spēcīgi sakrata. Ja nepieciešams, lai pilnīgi izšķīstu lipīdu fāze, liek mēģeni uz piecām minūtēm ūdens vannā, kur ir 60 °C. Ātri atdzesē tekošā ūdenī.
475.1.1.4. Izfiltrē daļu šķīduma ar membrānfiltru.
475.1.1.5. Filtrātam veic hromatogrāfiju tajā pašā dienā.
475.1.2. Parauga gatavošana, pievienojot iekšējo standartu:
475.1.2.1. Ar precizitāti līdz trešajai zīmei aiz komata stikla mēģenē iesver 1±0,1 g (a grami) parauga.
475.1.2.2. Pievieno 0,5 ml fosforskābes un 0,50 ml iekšējā standarta šķīduma un 9 ml etanola.
475.1.2.3. Noslēdz mēģeni un spēcīgi sakrata. Ja nepieciešams, lai pilnīgi izšķīstu lipīdu fāze, liek mēģeni uz piecām minūtēm ūdens vannā, kur ir 60 °C. Ātri atdzesē tekošā ūdenī.
475.1.2.4. Izfiltrē daļu šķīduma ar membrānfiltru.
475.1.2.5. Filtrāta hromatogrāfiju veic tajā pašā dienā.
475.2. Veicot gāzu hromatogrāfiju, ieteicami ir šādi darba apstākļi:
475.2.1. Kolonna:
475.2.1.1. Nerūsējošs tērauds.
475.2.1.2. Garums 2 m.
475.2.1.3. Diametrs 1/8''.
475.2.1.4. Pildījums 10 % SP™ 1000 (vai līdzvērtīgs) + 1 % H3PO4 uz Chromosorb WAW ar daļiņu izmēru 100–120.
475.2.2. Temperatūra:
475.2.2.1. Inžektors: 200 °C.
475.2.2.2. Kolonna: 120 °C.
475.2.2.3. Detektors: 200 °C.
475.2.2.4. Nesējgāze: slāpeklis.
475.2.2.5. Plūsma: 25 ml/min.
475.3. Hromatogrāfija:
475.3.1. Kalibrēšana:
475.3.1.1. Uz kalibrētām 20 ml kolbām ar pipeti attiecīgi pārnes 0,25, 0,50, 1,00, 2,00 un 4,00 ml propionskābes šķīduma (atbilstoši šā pielikuma 473.5.apakšpunktam).
475.3.1.2. Ar pipeti uz katru mērkolbu pārnes 1,00 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 473.6.apakšpunktam), uzpilda līdz zīmei ar etanolu un sajauc. Tā sagatavotajos šķīdumos ir e mg/ml 2-metilpropionskābes, kas ir iekšējais standarts (t.i., 1 mg/ml, ja e = 1000) un p/4, p/2, p, 2p, 4p mg/ml propionskābes (t.i., 0,25, 0,50, 1,00, 2,00, 4,00 mg/ml, ja p = 1000).
475.3.1.3. Ievada 1 µl katra šī šķīduma un, atzīmējot propionskābes/2-metilpropionskābes masas attiecību uz x ass un attiecīgo pīķu laukumu attiecību uz y ass, iegūst kalibrēšanas līkni.
475.3.1.4. Katru šķīdumu ievada trīs reizes un aprēķina vidējo pīķu laukumu attiecību.
475.3.2. Kvantitatīva noteikšana:
475.3.2.1. Ievada 1 µl parauga filtrāta (atbilstoši šā pielikuma 475.1.1.apakšpunktam).
475.3.2.2. Salīdzina hromatogrammu ar viena standarta šķīduma hromatogrammu (atbilstoši šā pielikuma 475.3.1.apakšpunktam). Ja kāda pīķa izdalīšanas laiks ir aptuveni vienāds ar 2-metilpropionskābei atbilstīgo izdalīšanas laiku, tad nomaina iekšējo standartu. Ja nav nekādu traucējošu faktoru, ievada 1 µl parauga filtrāta (atbilstoši šā pielikuma 475.1.2.apakšpunktam) un izmēra propionskābes pīķa un iekšējā standarta pīķa laukumu.
475.3.2.3. Katru šķīdumu ievada trīs reizes un aprēķina vidējo pīķu laukumu attiecību.
476. Aprēķinus veic šādā kārtībā:
476.1. No kalibrēšanas līknes, ko iegūst saskaņā ar šī pielikuma 475.3.1.apakšpunktu, nolasa masas (K) attiecību, kura atbilst pīķu laukumu attiecībai, ko aprēķina saskaņā ar šā pielikuma 475.3.2.apakšpunktu.
476.2. No tā iegūtās masas attiecības aprēķina propionskābes saturu paraugā (X) masas procentos pēc formulas:
x % (m/m) = K((0,5 × 100 × e)/(50 × a)) = K(e/a), kur
K – attiecība, ko aprēķina saskaņā ar šā pielikuma 476.1.apakšpunktu;
e – saskaņā ar šā pielikuma 473.6.apakšpunktu svērtā iekšējā standarta masa (gramos);
a – saskaņā ar šā pielikuma 475.1.2.apakšpunktu svērtā parauga masa (gramos).
Rezultātu noapaļo līdz vienai zīmei aiz komata.
477. No viena parauga ar 2 % (m/m) propionskābes saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt 0,12 %4.
37. Hidrohinona, hidrohinona monometilētera, hidrohinona monoetilētera un hidrohinona monobenzilētera kvalitatīva un kvantitatīva noteikšana kosmētikas līdzekļos
37.1. Hidrohinona, hidrohinona monometilētera, hidrohinona monoetilētera un hidrohinona monobenzilētera kvalitatīva noteikšana kosmētikas līdzekļos
478. Ar hidrohinona, hidrohinona monometilētera, hidrohinona monoetilētera un hidrohinona monobenzilētera kvalitatīvas noteikšanas metodi kvalitatīvi un kvantitatīvi nosaka hidrohinonu, hidrohinona monometilēteri, hidrohinona monoetilēteri un hidrohinona monobenzilēteri (monobenzonu) kosmētikas līdzekļos, kas paredzēti ādas balināšanai.
479. Hidrohinonu un tā ēterus kvalitatīvi nosaka plānslāņa hromatogrāfijā.
480. Hidrohinona un tā ēteru kvalitatīvas noteikšanas metodei izmanto šādus reaģentus1:
480.1. Etanols, 96 % (v/v).
480.2. Hloroforms.
480.3. Dietilēteris.
480.4. Attīstīstošais šķīdinātājs: Hloroforms/dietilēteris 66:33 (v/v).
480.5. Amonjaks, 25 % (m/m) (![]() = 0,91 g/ml).
= 0,91 g/ml).
480.6. Askorbīnskābe.
480.7. Hidrohinons.
480.8. Hidrohinona monometilēteris.
480.9. Hidrohinona monoetilēteris.
480.10. Hidrohinona monobenzilēteris (monobenzons).
480.11. Standartšķīdumi: standartšķīdumi būtu jāgatavo tieši pirms izmantošanas, un tie ir stabili vienu dienu:
480.11.1. Iesver 0,05 g hidrohinona 10 ml mēģenē ar iedaļām. Pievieno 0,250 g askorbīnskābes un 5 ml etanola. Pievieno amonjaku (atbilstoši šā pielikuma 480.5.apakšpunktam), līdz pH ir 10, un uzpilda līdz 10 ml tilpumam ar etanolu.
480.11.2. Iesver 0,05 g hidrohinona monometilētera 10 ml mēģenē ar iedaļām. Pievieno 0,250 g askorbīnskābes un 5 ml etanola. Pievieno amonjaku (atbilstoši šā pielikuma 480.5.apakšpunktam), līdz pH ir 10, un uzpilda līdz 10 ml tilpumam ar etanolu.
480.11.3. Iesver 0,05 g hidrohinona monoetilētera 10 ml mēģenē ar iedaļām. Pievieno 0,250 g askorbīnskābes un 5 ml etanola. Pievieno amonjaku (atbilstoši šā pielikuma 480.5.apakšpunktam), līdz pH ir 10, un uzpilda līdz 10 ml tilpumam ar etanolu.
480.11.4. Iesver 0,05 g hidrohinona monobenzilētera 10 ml mēģenē ar iedaļām. Pievieno 0,250 g askorbīnskābes un 5 ml etanola. Pievieno amonjaku (atbilstoši šā pielikuma 480.5.apakšpunktam), līdz pH ir 10, un uzpilda līdz 10 ml tilpumam ar etanolu.
480.12. Sudraba nitrāts.
480.13. 12-molibdofosforskābe.
480.14. Kālija fericianīda heksahidrāts.
480.15. Dzelzs(III) hlorīda heksahidrāts.
480.16. Aerosola reaģenti:
480.16.1. Pievieno 5 % (m/v) sudraba nitrāta ūdens šķīdumam amonjaku (atbilstoši šā pielikuma 480.5.apakšpunktam), līdz izšķīst nogulsnes, kas veidojas. Stāvot šķīdums kļūst sprādzienbīstams, un pēc izmantošanas tas jāizlej.
480.16.2. 10 % (m/v) 12-molibdofosforskābes šķīdums etanolā.
480.16.3. Sagatavo 1 % (m/v) kālija fericianīda ūdens šķīdumu un 2 % (m/v) dzelzs(III) hlorīda ūdens šķīdumu. Tieši pirms izmantošanas sajauc abus šķīdumus vienādās daļās.
481. Hidrohinona un tā ēteru kvalitatīvas noteikšanas metodei izmanto šādu iekārtu:
481.1. Parastais laboratorijas aprīkojums.
481.2. Parastais plānslāņa hromatogrāfijas aprīkojums.
481.3. Izmantošanai gatavas 20 × 20 cm silikagela GHR/UV254 (Machery, Nagel vai līdzvērtīgas) plānslāņa hromatogrāfijasplates. Slāņa biezums 0,25 mm.
481.4. Ultraskaņas vanna.
481.5. Centrifūga.
481.6. Ultravioleto staru lampa, 254 nm.
482. Hidrohinona un tā ēteru kvalitatīvu noteikšanu veic šādā kārtībā:
482.1. Parauga gatavošana:
482.1.1. Iesver 3,0 g parauga 10 ml mēģenē ar iedaļām.
482.1.2. Pievieno 0,250 g askorbīnskābes un 5 ml etanola.
482.1.3. Noregulē šķīduma pH uz 10 ar amonjaku (atbilstoši šā pielikuma 480.5.apakšpunktam).
482.1.4. Uzpilda līdz 10 ml tilpumam ar etanolu.
482.1.5. Noslēdz mēģeni ar aizbāzni un homogenizē ultraskaņas vannā 10 minūtes.
482.1.6. Izfiltrē caur filtrpapīru vai centrifugē ar ātrumu 3000 apgriezienu minūtē.
482.2. Plānslāņa hromatogrāfija:
482.2.1. Piesātina hromatogrāfijas kameru ar attīstīšanas šķīdinātāju (atbilstoši šā pielikuma 480.4.apakšpunktam).
482.2.2. Nogulsnē uz plates 2 µl standartšķīduma (atbilstoši šā pielikuma 480.11.apakšpunktam) un 2 µl parauga šķīduma (atbilstoši šā pielikuma 482.1.apakšpunktam). Attīsta tumsā apkārtējā temperatūrā, līdz šķīdinātāja fronte pavirzās par 15 cm no starta līnijas.
482.2.3. Izņem plati no kameras un ļauj žūt istabas temperatūrā.
482.3. Noteikšana:
482.3.1. Apskata plati 254 nm ultravioletā gaismā un atzīmē plankumus.
482.3.2. Apsmidzina plati ar sudraba nitrāta reaģentu (atbilstoši šā pielikuma 480.16.1.apakšpunktam) vai ar 12-molibdofosforskābes reaģentu (atbilstoši šā pielikuma 482.16.2.apakšpunktam) (karsē aptuveni līdz 120 °C), vai ar kālija fericianīda šķīdumu un dzelzs(III) hlorīda šķīdumu (atbilstoši šā pielikuma 482.16.3.apakšpunktam).
483. Kvalitatīvas noteikšanas aprēķinus veic šādā kārtībā:
483.1. Aprēķina katra plankuma Rf vērtību.
483.2. Salīdzina parauga šķīduma plankumus ar standartšķīdumu plankumiem, ņemot vērā to Rf vērtības, plankumu krāsu ultravioletajos staros un plankumu krāsu, kad tie padarīti redzami ar aerosola reaģentu.
483.3. Veic augstas izšķirtspējas plānslāņa hromatogrāfiju saskaņā ar šā pielikuma 37.2.apakšnodaļu un salīdzina parauga pīķa(-u) izdalīšanas laikus ar standartšķīdumu pīķu izdalīšanas laikiem.
483.4. Pēc plānslāņa hromatogrāfijasun augstas izšķirtspējas plānslāņa hromatogrāfijasrezultātiem kvalitatīvi nosaka hidrohinonu un (vai) tā ēterus.
484. Aprakstītajos apstākļos ir novērotas šādas Rf vērtības:
484.1. Hidrohinons: 0,32.
484.2. Hidrohinona monometilēteris: 0,53.
484.3. Hidrohinona monoetilēteris: 0,55.
484.4. Hidrohinona monobenziletilēteris: 0,58.
37.2. Hidrohinona, hidrohinona monometilētera, hidrohinona monoetilētera un hidrohinona monobenzilētera kvantitatīva noteikšana kosmētikas līdzekļos*
485. Ar hidrohinona, hidrohinona monometilētera, hidrohinona monoetilētera un hidrohinona monobenzilētera kvantitatīvas noteikšanas metodi kvantitatīvi nosaka hidrohinonu, hidrohinona monometilēteri, hidrohinona monoetilēteri un hidrohinona monobenzilēteri kosmētikas līdzekļos, kas paredzēti ādas balināšanai.
486. Paraugu ekstrahē ar ūdens/metanola maisījumu, viegli karsējot, lai izkūst visas lipīdu vielas. Analizējamās vielas iegūtajā šķīdumā kvantitatīvi nosaka apgrieztas fāzes šķidruma hromatogrāfijā ar ultravioleto staru detektoru.
487. Hidrohinona un tā ēteru kvantitatīvas noteikšanas metodei izmanto šādus reaģentus3:
487.1. Metanols.
487.2. Hidrohinons.
487.3. Hidrohinona monometilēteris.
487.4. Hidrohinona monoetilēteris.
487.5. Hidrohinona monobenzilēteris (monobenzons).
487.6. Tetrahidrofurāns augstas izšķirtspējas plānslāņa hromatogrāfijai.
487.7. Ūdens/metanola maisījums 1:1 (v/v): sajauc vienu tilpuma vienību ūdens un vienu tilpuma vienību metanola.
487.8. Kustīgā fāze: tetrahidrofurāna/ūdens maisījums 45:55 (v/v). Sajauc 45 tilpuma vienības tetrahidrofurāna un 55 tilpuma vienības ūdens.
487.9. Standartšķīdums: iesver 0,06 g hidrohinona, 0,08 g hidrohinona monometilētera, 0,10 g hidrohinona monoetilētera un 0,12 g hidrohinona monobenzilētera 50 ml mērkolbā. Izšķīdina un uzpilda līdz zīmei ar metanolu. Sagatavo standartšķīdumu, atšķaidot 10,00 ml šī šķīduma līdz 50,00 ml ar ūdens/metanola maisījumu (atbilstoši šā pielikuma 487.7.apakšpunktam). Šie šķīdumi jāgatavo tieši pirms izmantošanas.
488. Hidrohinona un tā ēteru kvalitatīvas noteikšanas metodei izmanto šādu iekārtu:
488.1. Parastais laboratorijas aprīkojums.
488.2. Ūdens vanna, kurā var uzturēt 60 °C temperatūru.
488.3. Augstas izšķirtspējas šķidruma hromatogrāfs ar maināma viļņu garuma ultravioleto staru detektoru un 10 µl ievadīšanas cilpu.
488.4. Hromatogrāfijas kolonna: nerūsējoša tērauda hromatogrāfijas kolonna, kuras garums ir 250 mm, iekšējais diametrs 4,6 mm, pildījums Zorbax fenils (ķīmiski saistīts fenetilsilāns uz Zorbax SIL, kam atlikušas ar trimetilhlorsilānu inaktivētas silolgrupas), kura daļiņu izmērs ir 6 µm, vai līdzvērtīgs. Neizmanto aizsargkolonnu, izņemot fenila vai līdzvērtīgu.
488.5. Filtrpapīrs ar 90 mm diametru, Schleicher & Schull, Weissband No 5892 vai līdzvērtīgs.
489. Hidrohinona un tā ēteru kvalitatīvu noteikšanu veic šādā kārtībā:
489.1. Parauga gatavošana:
489.1.1. Ar precizitāti līdz trešajai zīmei aiz komata 50 ml mērkolbā iesver 1±0,1 g (a grami) parauga.
489.1.2. Disperģē paraugu 25 mililitros ūdens/metanola maisījuma (atbilstoši šā pielikuma 487.7.apakšpunktam).
489.1.3. Noslēdz kolbu un spēcīgi krata, līdz rodas vienmērīga suspensija. Krata vismaz vienu minūti.
489.1.4. Liek kolbu ūdens vannā, kurā uztur 60 °C, lai paātrinātu ekstrakciju.
489.1.5. Atdzesē kolbu un uzpilda līdz atzīmei ar ūdeni/metanolu (atbilstoši šā pielikuma 487.7.apakšpunktam).
489.1.6. Izfiltrē ekstraktu caur filtrpapīru.
489.1.7. Pēc ekstrakta sagatavošanas 24 stundās veic kvantitatīvu noteikšanu augstas izšķirtspējas plānslāņa hromatogrāfijā.
489.2. Augstas izšķirtspējas šķidruma hromatogrāfija:
489.2.1. Kustīgās fāzes (atbilstoši šā pielikuma 487.8.apakšpunktam) plūsmas ātrumu noregulē uz 1,0 ml/min un detektora viļņu garumu uz 295 nm.
489.2.2. Ievada 10 µl parauga šķīduma, ko iegūst saskaņā ar šā pielikuma 489.1.apakšpunktu, un uzņem hromatogrammu. Izmēra pīķu laukumus. Kalibrē saskaņā ar šā pielikuma 489.2.3.apakšpunktu. Salīdzina parauga un standartšķīdumu hromatogrammas. Pēc pīķu laukumiem un relatīvās signāla attiecības pret masas vienību (Rf), ko aprēķina saskaņā ar šā pielikuma 489.2.3.apakšpunktu, aprēķina analizējamo vielu koncentrāciju parauga šķīdumā.
489.2.3. Kalibrēšana:
489.2.3.1. Ievada 10 µl standartšķīduma (atbilstoši šā pielikuma 487.9.apakšpunktam) un uzņem hromatogramu. Ievada vairākas reizes, līdz iegūst nemainīgu pīķu laukumu.
489.2.3.2. Nosaka relatīvo signāla attiecību pret masas vienību RFi:
RFi = pi/ci, kur
pi – hidrohinona, hidrohinona monometilētera, hidrohinona monoetilētera vai hidrohinona monobenzilētera pīķu laukums;
ci – hidrohinona, hidrohinona monometilētera, hidrohinona monoetilētera vai hidrohinona monobenzilētera koncentrācija (g/50 ml) standartšķīdumā (atbilstoši šā pielikuma 487.9.apakšpunktam).
489.2.3.3. Nosaka, vai iegūtās standartšķīduma un parauga šķīduma hromatogrammas atbilst šādām prasībām:
489.2.3.3.1. Minimālais divu pīķu atdalījums ir vismaz 0,90 (pīķu atdalījums definēts šī pielikuma 10.attēlā).
10.attēls
Pīķu atdalījums
Ja nav panākts vajadzīgais atdalījums, būtu vai nu jāizmanto efektīvāka kolonna, vai arī jāregulē kustīgās fāzes sastāvs, līdz panākta atbilstība prasībām.
489.2.3.3.2. Visu iegūto pīķu asimetrijas faktors AS ir 0,9–1,5 (pīķu asimetrijas faktors definēts šī pielikuma 11.attēlā). Lai uzņemtu hromatogrammu asimetrijas faktora noteikšanai, ieteicamais pašrakstītāja ātrums ir vismaz 2 cm minūtē.
11.attēls
Pīķu asimetrijas faktors
489.2.3.3.3. Iegūst stabilu bāzes līniju.
490. Pēc analizējamo vielu pīķu laukumiem aprēķina analizējamās(-o) vielas(-u) koncentrāciju paraugā. Aprēķina analizējamās vielas koncentrāciju paraugā masas procentos (xi) pēc formulas:
xi % (m/m) = (bi × 100)/(RFi × a), kur
a – parauga masa (gramos);
bi – paraugā esošās analizējamās vielas i pīķa laukums.
491. Atkārtojamību izvērtē šādi4:
491.1. No viena parauga ar aptuveni 2,0 % hidrohinona saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,13 % vērtību.
491.2. No viena parauga ar aptuveni 1,0 % hidrohinona monometilētera saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,1 % vērtību.
491.3. No viena parauga ar aptuveni 1,0 % hidrohinona monoetilētera saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,11 % vērtību.
491.4. No viena parauga ar aptuveni 1,0 % hidrohinona monobenzilētera saturu divās paralēlās kvantitatīvās noteikšanās iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,11 % vērtību.
492. Reproducējamību izvērtē šādi4:
492.1. No viena parauga ar 2,0 % hidrohinona saturu divās paralēlās kvantitatīvās noteikšanās atšķirīgos apstākļos (citas laboratorijas, citi laboranti, citas iekārtas un (vai) cits laiks) iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,37 % vērtību.
492.2. No viena parauga ar 1,0 % hidrohinona monometilētera saturu divās paralēlās kvantitatīvās noteikšanās atšķirīgos apstākļos (citas laboratorijas, citi laboranti, citas iekārtas un (vai) cits laiks) iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,21 % vērtību.
492.3. No viena parauga ar 1,0 % hidrohinona monoetilētera saturu divās paralēlās kvantitatīvās noteikšanās atšķirīgos apstākļos (citas laboratorijas, citi laboranti, citas iekārtas un (vai) cits laiks) iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,19 % vērtību.
492.4. No viena parauga ar 1,0 % hidrohinona monobenzilētera saturu divās paralēlās kvantitatīvās noteikšanās atšķirīgos apstākļos (citas laboratorijas, citi laboranti, citas iekārtas un (vai) cits laiks) iegūto rezultātu starpībai nevajadzētu pārsniegt absolūtu 0,11 % vērtību.
*Piezīmes.
1. Ja hidrohinona saturs izrādās ievērojami augstāks nekā 2 % un tā saturs jāprognozē precīzi, parauga ekstrakts (atbilstoši šā pielikuma 489.1.apakšpunktam) būtu jāatšķaida līdz tādai koncentrācijai, kas ir līdzīga koncentrācijai, kuru iegūtu no parauga, kas satur 2 % hidrohinona, un kvantitatīvā noteikšana būtu jāatkārto (dažās iekārtās absorbcija var būt ārpus detektora lineārā diapazona, ja hidrohinona koncentrācija ir augsta).
2. Ar minēto metodi var kvantitatīvi noteikt hidrohinonu un tā ēterus vienā izokrātiskā analīzē. Fenila kolonna nodrošina pietiekamu hidrohinona izdalīšanu, ko nevar garantēt, izmantojot C18 kolonnu ar minēto kustīgo fāzi. Tomēr šī metode nenovērš traucējumus, ko izraisa vairāki parabeni. Tādā gadījumā kvantitatīvo noteikšanu atkārto ar citu kustīgās fāzes/nekustīgās fāzes sistēmu. Piemērotas metodes ir norādītas šādos avotos:
2.1. M.Herpol-Borremans et M.-O. Masse, Identification et dosage de l'hydroquinone et de ses éthers méthylique et benzylique dans les produits cosmétiques pour blanchir la peau. Int. j. Cosmet. Sci. 8-203-214 (1986):
2.1.1. kolonna: Zorbax ODS, 4,6 × 25 mm vai līdzvērtīga;
2.1.2. temperatūra 36 °C;
2.1.3. plūsma 1,5 ml/min;
2.1.4. kustīgā fāze: hidrohinonam – metanols/ūdens 5:95 (v/v), hidrohinona monometilēterim – metanols/ūdens 30:70 (v/v), hidrohinona monobenzilēterim – metanols/ūdens 80:20 (v/v);
2.2. Apstākļi, piemēroti hidrohinonam: J. Firth and I. Rix, Determination of hydroquinone in skin toning creams, Analyst (1986), 111, p. 129.:
2.2.1. kolonna: Spherisorb S5-ODS vai līdzvērtīga;
2.2.2. kustīgā fāze: ūdens/metanols 90:10 (v/v);
2.2.3. plūsma 1,5 ml/min.
38. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta pierādīšana un noteikšana kosmētikas līdzekļos
38.1. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta pierādīšana
493. Ar šajā nodaļā noteikto metodi pierāda 2-fenoksietanolu, 1-fenoksipropān-2-olu, metil-4-hidroksibenzoātu, etil-4-hidroksibenzoātu, propil-4-hidroksibenzoātu, butil-4-hidroksibenzoātu un benzil-4-hidroksibenzoātu kosmētikas līdzekļos.
494. Konservantus no paskābinātā kosmētikas līdzekļa parauga ekstrahē ar acetonu. Pēc filtrēšanas acetona šķīdumu sajauc ar ūdeni un sārmainā vidē nogulsnē taukskābes to kalcija sāļu veidā. Sārma acetona/ūdens maisījumu ekstrahē ar dietilēteri, lai atdalītu lipofilas vielas. Pēc paskābināšanas konservantus ekstrahē ar dietilēteri. Dietilētera ekstrakta alikvoto daļu uznes uz silikagela plānslāņa plates. Pēc plates attīstīšanas iegūto hromatogrammu apskata ultravioletā gaismā un padara redzamu ar Milona reaģentu.
495. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta pierādīšanas metodei izmanto šādus reaģentus1, 3:
495.1. Acetons.
495.2. Dietilēteris.
495.3. n-pentāns.
495.4. Metanols.
495.5. Ledus etiķskābe.
495.6. Sālsskābes šķīdums, c(HCl) = 4 mol/l.
495.7. Kālija hidroksīda šķīdums, c(KOH) = 4 mol/l.
495.8. Kalcija hlorīda dihidrāts (CaCl2·2H2O).
495.9. Noteikšanas reaģents: Milona reaģents (Milona reaģents (dzīvsudraba(II) nitrāts) ir nopērkams lietošanai gatavs šķīdums (Fluka 69820)).
495.10. 2-fenoksietanols.
495.11. 1-fenoksipropān-2-ols.
495.12. Metil-4-hidroksibenzoāts (metilparabens).
495.13. Etil-4-hidroksibenzoāts (etilparabens).
495.14. n-propil-4-hidroksibenzoāts (propilparabens).
495.15. n-butil-4-hidroksibenzoāts (butilparabens).
495.16. Benzil-4-hidroksibenzoāts (benzilparabens).
495.17. Standartšķīdumi: no katras standartvielas (atbilstoši šā pielikuma 495.11., 495.12., 495.13., 495.14., 495.15., 495.16. un 495.17.apakšpunktam) gatavo 0,1 % (m/v) šķīdumu metanolā.
495.18. Attīstīšanas šķīdinātājs: sajauc 88 tilpuma vienības n-pentāna ar 12 tilpuma vienībām ledus etiķskābes.
496. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta pierādīšanas metodei izmanto laboratorijas standartaprīkojumu un šādu aprīkojumu:
496.1. Ūdens vanna, kurā var uzturēt 60 °C.
496.2. Attīstīšanas kamera (kas nav izklāta ar filtrpapīru).
496.3. Ultravioletas gaismas avots, 254 nm.
496.4. Plānslāņa plates, 20 × 20 cm, kas iepriekš pārklātas ar 0,25 mm silikagelu 60F254, ar koncentrēšanas zonu (Merck Nr 11798, Darmstadt vai līdzvērtīgas).
496.5. Krāsns, kurā var uzturēt 105 °C.
496.6. Karsta gaisa fēns.
496.7. Vilnas krāsu rullītis, apmēram 10 cm garš, ar ārējo diametru aptuveni 3,5 cm. Vilnas kārtas biezums ir 2–3 mm. Ja nepieciešams, vilnu apcērp.
496.8. Stikla mēģenes, 50 ml, ar skrūvējamu vāciņu.
496.9. Elektriskā plītiņa ar termostatu temperatūras regulēšanai. Temperatūras uzstādījums: ap 80 °C. Elektrisko plītiņu pārklāj ar apmēram 6 mm biezu 20 × 20 cm alumīnija plati, lai nodrošinātu vienmērīgu sasilšanu.
497. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta pierādīšanu veic šādā kārtībā:
497.1. Parauga gatavošana:
497.1.1. Iesver aptuveni 1 g parauga 50 ml stikla mēģenē ar skrūvējamu vāciņu. Pievieno četrus pilienus sālsskābes šķīduma (atbilstoši šā pielikuma 495.7.apakšpunktam) un 40 ml acetona.
497.1.2. Izteikti bāziskiem kosmētikas līdzekļiem (piemēram, tualetes ziepēm) pievieno 20 pilienus sālsskābes šķīduma.
497.1.3. Aizskrūvē mēģeni, viegli karsē maisījumu aptuveni līdz 60 °C, lai paātrinātu konservantu ekstrakciju acetona fāzē, un stipri krata vienu minūti.
497.1.4. Izmēra šķīduma pH ar indikatorpapīru un ar sālsskābes šķīdumu noregulē šķīduma pH uz 3.
497.1.5. Atkal stipri krata vienu minūti.
497.1.6. Atdzesē šķīdumu līdz istabas temperatūrai un izfiltrē caur filtrpapīru koniskā kolbā.
497.1.7. Pārnes 20 ml filtrāta uz 200 ml konisku kolbu, pievieno 60 ml ūdens un sajauc.
497.1.8. Ar kālija hidroksīdu (atbilstoši šā pielikuma 495.7.apakšpunktam), lietojot pH indikatorpapīru, noregulē maisījuma pH aptuveni uz 10.
497 1.9. Pievieno 1 g kalcija hlorīda dihidrāta un stipri sakrata.
497.1.10. Izfiltrē šķīdumu caur filtrpapīru 250 ml dalāmajā piltuvē, kur ir 75 ml dietilētera, un stipri krata vienu minūti.
497.1.11. Ļauj fāzēm atdalīties un savāc ūdens slāni koniskā 200 ml kolbā.
497.1.12. Ar sālsskābes šķīdumu, lietojot pH indikatorpapīru, noregulē šķīduma pH aptuveni uz 2.
497.1.13. Pēc tam pievieno 10 ml dietilētera un stipri krata vienu minūti.
497.1.14. Ļauj fāzēm atdalīties un pārnes aptuveni 2 ml dietilētera slāņa uz 5 ml parauga mēģeni.
497.2. Plānslāņa hromatogrāfija (TLC):
497.2.1. Noliek TLC plati uz sakarsētās alumīnija plates.
497.2.2. Uznes 10 μl katra standartšķīduma (atbilstoši šā pielikuma 495.17.apakšpunktam) un 100 µl parauga šķīduma(u) (atbilstoši šā pielikuma 497.1.apakšpunktam) uz starta līnijas TLC plates koncentrēšanas zonā. Pēc vēlēšanās šķīdinātāja iztvaikošanu var paātrināt ar gaisa plūsmu.
497.2.3. Noņem TLC plati no plītiņas un ļauj atdzist līdz istabas temperatūrai. Pārnes 100 ml attīstīšanas šķīdinātāja (atbilstoši šā pielikuma 495.18.apakšpunktam) uz attīstīšanas kameru.
497.2.4. Tūlīt liek TLC plati nepiesātinātajā kamerā un attīsta istabas temperatūrā, līdz šķīdinātāja fronte ir pavirzījusies apmēram 15 cm no sākuma līnijas.
497.2.5. Izņem plati no attīstīšanas kameras un žāvē karsta fēna gaisa plūsmā.
497.2.6. Apskata plati ultravioletā gaismā un atzīmē plankumu izvietojumu.
497.2.7. Karsē plati 30 minūtes krāsnī 100 °C, lai atdalītu etiķskābes pārākumu. Konservantus hromatogrammā padara redzamus ar Milona reaģentu (atbilstoši šā pielikuma 495.9.apakšpunktam), tajā iegremdējot krāsu rullīti (atbilstoši šā pielikuma 496.7.apakšpunktam) un ar to uznesot reaģentu uz TLCplates, līdz plate ir vienmērīgi samitrināta. Alternatīvi plankumus var padarīt redzamus, uzmanīgi uzpilinot Milona reaģentu uz katra ultravioletajā gaismā atzīmētā plankuma. 4-hidroksibenzoskābes esteri parādās sarkanu plankumu veidā, 2-fenoksietanols un 1-fenoksipropān-2-ols – dzeltenu plankumu veidā. Tomēr pati 4-hidroksibenzoskābe, kas var būt kā konservants vai parabenu sadalīšanās produkts paraugos, arī parādās sarkana plankuma veidā*.
497.3. Pierādīšana:
497.3.1. Aprēķina katra plankuma Rf vērtību.
497.3.2. Salīdzina no parauga šķīduma iegūtos plankumus ar tiem, kas iegūti no standartšķīdumiem, salīdzinot to Rf vērtības, to raksturu ultravioletajos staros un krāsu pēc tam, kad tie ir padarīti redzami.
497.3.3. Izdara provizoriskus secinājumus par konservantu identitāti.
Parabenu klātbūtnes gadījumā būtu jāveic šī pielikuma 38.2.apakšnodaļā minētā HPLC procedūra. Apvienojot TLC un augstas izšķirtspējas šķidruma hromatogrāfijas (HPLC) rezultātus, apstiprina 2-fenoksietanola, 1-fenoksipropān-2-ola un parabenu klātbūtni.
* Piezīmes.
1. Tā kā Milona reaģents ir toksisks, labāk to lietot saskaņā ar vienu no minētajām procedūrām. Smidzināt nav ieteicams.
2. Arī citi savienojumi, kuros ir hidroksilgrupas, var krāsoties saskarsmē ar Milona reaģentu. To krāsu un Rf vērtību tabula, ko iegūst no vairākiem konservantiem, izmantojot šo TLC procedūru, ir atrodama N.deKruijf, M.A.H.Rijk, L.A.Pranato-Soetardhi and A.Schouten (1987): Determination of preservatives in cosmetic products I: Thin-layer chromatographic procedure for the identification of preservatives in cosmetic products (J.Chomatography 410, 395-411).
3. Šajā tabulā minētās Rf vērtības norāda vērtības, kādas var iegūt:
|
Savienojums |
hRf |
Krāsa |
|
4-hidroksibenzoskābe |
11 |
sarkana |
|
metilparabens |
12 |
sarkana |
|
etilparabens |
17 |
sarkana |
|
propilparabens |
21 |
sarkana |
|
butilparabens |
26 |
sarkana |
|
benzilparabens |
16 |
sarkana |
|
2-fenoksietanols |
29 |
dzeltena |
|
1-fenoksipropān-2-ols |
50 |
dzeltena |
4. Nav atšķirības starp 4-hidroksibenzoskābi un metilparabenu vai benzilparabenu un etilparabenu. Šo savienojumu pierādīšanu apstiprina ar šī pielikuma 38.2.apakšnodaļā minēto HPLC metodi un salīdzinot parauga un standartu izdalīšanas laikus.
38.2. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta noteikšana*
498. Ar 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta noteikšanas metodi kosmētikas līdzekļos nosaka 2-fenoksietanolu, 1-fenoksipropān-2-olu, metil-4-hidroksibenzoātu, etil-4-hidroksibenzoātu, propil-4-hidroksibenzoātu, butil-4-hidroksibenzoātu un benzil-4-hidroksibenzoātu.
499. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta (konservantu) daudzumu izsaka masas procentos.
500. Paraugu paskābina, pievienojot sērskābi, un pēc tam suspendē etanola un ūdens maisījumā. Maisījumu viegli karsē, lai izkausētu lipīdu fāzi un veicinātu kvantitatīvu ekstrakciju, pēc tam maisījumu izfiltrē. Konservantus filtrātā nosaka apgrieztas fāzes HPLC, par iekšējo standartu izmantojot izopropil-4-hidroksibenzoātu.
501. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta noteikšanas metodei izmanto šādus reaģentus1, 2, 3:
501.1. Absolūtais etanols.
501.2. 2-fenoksietanols.
501.3. 1-fenoksipropān-2-ols.
501.4. Metil-4-hidroksibenzoāts (metilparabens).
501.5. Etil-4-hidroksibenzoāts (etilparabens).
501.6. n-propil-4-hidroksibenzoāts (propilparabens).
501.7. Izopropil-4-hidroksibenzoāts (izopropilparabens).
501.8. n-butil-4-hidroksibenzoāts (butilparabens).
501.9. Benzil-4-hidroksibenzoāts (benzilparabens).
501.10. Tetrahidrofurāns.
501.11. Metanols.
501.12. Acetonitrils.
501.13. Sērskābes šķīdums, c(H2SO4) = 2 mol/l.
501.14. Etanola/ūdens maisījums: sajauc deviņas tilpuma vienības etanola un vienu tilpuma vienību ūdens.
501.15. Iekšējā standarta šķīdums: precīzi nosver aptuveni 0,25 g izopropilparabena, pārnes uz 500 ml mērkolbu, izšķīdina un uzpilda līdz zīmei ar etanola/ūdens maisījumu (atbilstoši šā pielikuma 501.14.apakšpunktam).
501.16. Kustīgā fāze: tetrahidrofurāna/ūdens/metanola/acetonitrila maisījums. Sajauc 5 tilpuma vienības tetrahidrofurāna, 60 tilpuma vienības ūdens, 10 tilpuma vienības metanola un 25 tilpuma vienības acetonitrila.
501.17. Konservanta standartšķīdums: precīzi iesver aptuveni 0,2 g 2-fenoksietanola, 0,2 g 1-fenoksipropān-2-ola, 0,05 g metilparabena, 0,05 g etilparabena, 0,05 g propilparabena, 0,05 g butilparabena un 0,025 g benzilparabena 100 ml mērkolbā, izšķīdina un uzpilda līdz atzīmei ar etanola/ūdens maisījumu. Glabāts ledusskapī, šķīdums saglabājas stabils vienu nedēļu.
501.18. Konservantu standartšķīdumi: attiecīgi pārnes 20,00, 10,00, 5,00, 2,00 un 1,00 ml standartšķīduma (atbilstoši šā pielikuma 501.17.apakšpunktam) uz 50 ml mērkolbām. Katrā kolbā pievieno 10,00 ml iekšējā standarta šķīduma (atbilstoši šā pielikuma 501.15.apakšpunktam) un 1,0 ml sērskābes šķīduma (atbilstoši šā pielikuma 501.13.apakšpunktam) un uzpilda līdz zīmei ar etanola/ūdens maisījumu. Šie šķīdumi būtu jāgatavo tieši pirms lietošanas.
502. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta noteikšanas metodei izmanto laboratorijas standartaprīkojumu un šādu aprīkojumu:
502.1. Ūdens vanna, kurā var uzturēt 60±1 °C.
502.2. Augstas izšķirtspējas šķidruma hromatogrāfs ar 280 nm ultraīsviļņu detektoru.
502.3. Hromatogrāfijas kolonna ar šādiem nosacījumiem:
502.3.1. Nerūsējošs tērauds.
502.3.2. Kolonnas garums 25 cm, iekšējais diametrs 4,6 mm (vai attiecīgi 12,5 cm un 4,6 mm).
502.3.3. Nucleosil 5C18 vai līdzvērtīgs pildījums.
502.4. Stikla mēģenes, 100 ml, ar skrūvējamu vāciņu.
502.5. Vārķermeņi, 2–4 mm karborunds vai līdzvērtīgi.
503. 2-Fenoksietanola, 1-fenoksipropān-2-ola, metil-, etil-, propil-, butil- un benzil-4-hidroksibenzoāta noteikšanu veic šādā kārtībā:
503.1. Parauga gatavošana:
503.1.1. Parauga gatavošana, nepievienojot iekšējo standartu:
503.1.1.1. Iesver aptuveni 1,0 g parauga 100 ml stikla mēģenē ar skrūvējamu vāciņu.
503.1.1.2. Ar pipeti mēģenē iepilina 1,0 ml sērskābes šķīduma (atbilstoši šā pielikuma 501.13.apakšpunktam) un 50,0 ml etanola/ūdens maisījuma (atbilstoši šā pielikuma 501.14.apakšpunktam).
503.1.1.3. Pievieno aptuveni 1 g vārķermeņu, noslēdz mēģeni un spēcīgi krata, līdz rodas vienmērīga suspensija. Krata vismaz vienu minūti.
503.1.1.4. Liek mēģeni uz piecām minūtēm ūdens vannā, kur uztur 60±1 °C, lai paātrinātu konservantu ekstrakciju etanola fāzē.
503.1.1.5. Tūlīt atdzesē mēģeni auksta ūdens strūklā un liek ekstraktu ledusskapī uz vienu stundu.
503.1.1.6. Izfiltrē ekstraktu caur filtrpapīru.
503.1.1.7. Pārnes aptuveni 2 ml filtrāta uz 5 ml parauga mēģeni.
503.1.1.8. Liek ekstraktus ledusskapī un 24 stundās veic noteikšanu HPLC.
503.1.2. Parauga gatavošana, pievienojot iekšējo standartu:
503.1.2.1. Iesver ar precizitāti līdz trim zīmēm aiz komata 1,0±0,1 g parauga 100 ml stikla mēģenē ar skrūvējamu vāciņu.
503.1.2.2. Ar pipeti mēģenē iepilina 1,0 ml sērskābes šķīduma un 40,0 ml etanola/ūdens maisījuma.
503.1.2.3. Pievieno aptuveni 1 g vārķermeņu un precīzi 10,00 ml iekšējā standarta šķīduma.
503.1.2.4. Noslēdz mēģeni un spēcīgi krata, līdz iegūst vienmērīgu suspensiju. Krata vismaz vienu minūti.
503.1.2.5. Liek mēģeni uz piecām minūtēm ūdens vannā, kur uztur 60±1 °C, lai paātrinātu konservantu ekstrakciju etanola fāzē.
503.1.2.6. Tūlīt atdzesē mēģeni auksta ūdens strūklā un liek ekstraktu ledusskapī uz vienu stundu.
503.1.2.7. Izfiltrē ekstraktu caur filtrpapīru.
503.1.2.8. Pārnes aptuveni 2 ml filtrāta uz 5 ml parauga mēģeni (parauga šķīdums).
503.1.2.9. Liek ekstraktu ledusskapī un 24 stundās veic noteikšanu HPLC.
503.2. Augstas izšķirtspējas šķidruma hromatogrāfija (HPLC):
503.2.1. Hromatogrāfijas apstākļi:
503.2.1.1. Kustīgā fāze: tetrahidrofurāna/ūdens/metanola/acetonitrila maisījums (atbilstoši šā pielikuma 501.16.apakšpunktam).
503.2.1.2. Plūsmas ātrums: 1,5 ml/minūtē.
503.2.1.3. Noteikšanas viļņa garums: 280 nm.
503.2.2. Kalibrēšana:
503.2.2.1. Ievada 10 µl katra konservanta standartšķīduma (atbilstoši šā pielikuma 501.18.apakšpunktam).
503.2.2.2. Pēc iegūtajām hromatogrammām nosaka konservantu standartšķīdumu pīķu augstumu attiecības pret iekšējā standarta pīķa augstumu.
503.2.2.3. Konstruē katra konservanta līkni, attiecinot minētās attiecības pret standartšķīdumu koncentrācijām.
503.2.3. Noteikšana:
503.2.3.1. Ievada 10 µl parauga šķīduma bez iekšējā standarta (atbilstoši šā pielikuma 503.1.1.apakšpunktam) hromatogrāfā un uzņem hromatogrammu.
503.2.3.2. Ievada 10 μl viena konservanta standarta šķīduma (atbilstoši šā pielikuma 501.18.apakšpunktam) un uzņem hromatogrammu.
503.2.3.3. Salīdzina iegūtās hromatogrammas: ja parauga ekstrakta (atbilstoši šā pielikuma 503.1.1.4.apakšpunktam) hromatogrammā nav neviena pīķa, kura izdalīšanas laiks ir aptuveni vienāds ar izopropilparabena (ieteicamais iekšējais standarts) pīķa izdalīšanas laiku, ievada 10 µl parauga šķīduma ar iekšējo standartu (atbilstoši šā pielikuma 503.1.2.apakšpunktam). Uzņem hromatogrammu un izmēra pīķu augstumus. Ja parauga šķīduma hromatogrammā ir traucējošs pīķis, kura izdalīšanas laiks ir aptuveni vienāds ar izopropilparabena pīķa izdalīšanas laiku, izvēlas citu iekšējo standartu. Ja kāds analizējamais konservants neparādās parauga hromatogrammā, šo konservantu var izmantot par alternatīvu iekšējo standartu.
503.2.3.4. Aprēķina analizējamo konservantu pīķu augstumu attiecības pret iekšējā standarta pīķa augstumu.
503.2.3.5. Pārliecinās, vai kalibrēšanā izmantoto standartšķīdumi veido taisnu līniju.
503.2.3.6. Pārliecinās, vai iegūtās standartšķīduma un parauga šķīduma hromatogrammas atbilst šādiem nosacījumiem:
503.2.3.6.1. Vissliktāk atdalīto pīķu pāra atdalījums ir vismaz 0,90 (pīķu atdalījums definēts šī pielikuma 12.attēlā).
|
Pīķu atdalījums (p) p = f/g |
 |
12.attēls
Pīķu atdalījums
Ja nav panākts vajadzīgais
atdalījums, būtu jālieto efektīvāka kolonna vai jāregulē mobilās fāzes sastāvs,
līdz panāk vajadzīgo atdalījumu
503.2.3.6.2. Asimetrijas faktors: visi iegūtie pīķi ir 0,9–1,5 (pīķu asimetrijas faktors definēts šī pielikuma 13.attēlā). Lai uzņemtu hromatogrammu asimetrijas faktora noteikšanai, ieteicams pašrakstītāja ātrums vismaz 2 cm minūtē.
|
Asimetrijas faktors (As) As = b/a |
|
13.attēls
Pīķu asimetrijas faktors
503.2.3.6.3. Iegūst vienmērīgu bāzes līniju.
504. Konservantu koncentrāciju parauga šķīdumā aprēķina pēc kalibrēšanas līknes (atbilstoši šā pielikuma 503.2.2.apakšpunktam) un analizējamo konservantu pīķu augstumu attiecībām pret iekšējā standarta pīķa augstumu. Aprēķina 2-fenoksietanola, 1-fenoksipropān-2-ola, metil-4-hidroksibenzoāta, etil-4-hidroksibenzoāta, propil-4-hidroksibenzoāta, butil-4-hidroksibenzoāta un benzil-4-hidroksibenzoāta saturu wi masas procentos (% m/m) pēc formulas:
% wi (m/m) = ((bi)/(200 × a)), kur
bi – konservanta i koncentrācija (µg/ml) parauga šķīdumā, ko nolasa no kalibrēšanas līknes;
a – analizējamā daudzuma masa (gramos).
505. Atkārtojamības4 un reproducējamības4 izvērtēšanas metode noteikta piezīmju 5.punktā.
* Piezīmes.
1. Nekustīgā fāze. Vielu izdalīšana, nosakot HPLC, lielā mērā ir atkarīga no nekustīgās fāzes veida, markas un izcelsmes. Par to, vai kolonnu var lietot analizējamo konservantu atdalīšanai, var spriest pēc rezultātiem, ko iegūst, hromatografējot standartšķīdumus (atbilstoši šā pielikuma 503.2.3.apakšpunktam). Papildus ieteiktajam Hypersil ODS kolonnas pildījumam par piemērotu ir atzīts arī Zorbax ODS. Alternatīvi var optimizēt ieteicamo kustīgās fāzes sastāvu, lai panāktu vajadzīgo atdalījumu.
2. Noteikšanas viļņa garums. Šo rādītāju analīzes laikā uzmanīgi kontrolē, jo aptuveni pārbaudot minēto metodi, ir noskaidrots, ka nelielas noteikšanas viļņa garuma izmaiņas var ievērojami ietekmēt noteikšanas rezultātus.
3. Traucējumi. Apstākļos, kas aprakstīti šajā metodē, eluē arī daudzus citus savienojumus, piemēram, konservantus un kosmētikas piedevas. Konservantu izdalīšanas laiki ir iekļauti N.deKruijf, M.A.H.Rijk, L.A.Pranato-Soetardhi and A.Schouten, (1989). Determination of preservatives in cosmetic products II. High-performance liquid chromatographic identification (J.Chromatography 469, 317-398).
4. Hromatogrāfijas kolonnu var aprīkot ar piemērotu aizsargkolonnu.
5. Metode ir pārbaudīta salīdzināmās pārbaudēs, piedaloties deviņām laboratorijām. Tika analizēti trīs paraugi. Šajā tabulā ir iekļauti katrā paraugā nosakāmo komponenšu vidējie % m/m (m), atkārtojamība (r) un reproducējamība (R):
|
Paraugs |
2-fenoksi-propān-2-ols |
1-fenoksi-propān-2-ols |
Metilparabens |
Etilparabens |
Propilparabens |
Butilparabens |
Benzilparabens |
|
|
Vitaminizēts krēms |
m |
1,124 |
0,250 |
0,0628 |
0,031 |
0,0906 |
||
|
r |
0,016 |
0,018 |
0,0035 |
0,0028 |
0,0044 |
|||
|
R |
0,176 |
0,030 |
0,0068 |
0,0111 |
0,0034 |
|||
|
Krēms, kas ātri iesūcas |
m |
1,196 |
0,266 |
0,076 |
||||
|
r |
0,040 |
0,003 |
0,002 |
|||||
|
R |
0,147 |
0,022 |
0,004 |
|||||
|
Masāžas krēms |
m |
0,806 |
0,180 |
0,148 |
0,152 |
|||
|
r |
0,067 |
0,034 |
0,013 |
0,015 |
||||
|
R |
0,112 |
0,078 |
0,012 |
0,016 |
||||
Piezīmes.
1 Visiem reaģentiem jābūt ar analītisku kvalitāti.
2 Visiem reaģentiem jābūt attiecīgos gadījumos piemērotiem HPLC.
3 Jāizmanto destilēts ūdens vai vismaz līdzvērtīgas kvalitātes ūdens.
4 Metožu pareizību novērtē atbilstoši šādiem standartiem:
1) LVS ISO 5725-1:2006 "Mērīšanas metožu un rezultātu pareizība (patiesums un precizitāte). 1.daļa: Vispārīgie principi un definīcijas.";
2) LVS ISO 5725-2:2006 "Mērīšanas metožu un rezultātu pareizība (patiesums un precizitāte). 2.daļa: Pamatmetode mērīšanas standartmetodes atkārtojamības un reproducējamības noteikšanai.";
3) LVS ISO 5725-3:2006 "Mērīšanas metožu un rezultātu pareizība (patiesums un precizitāte). 3.daļa: Mērīšanas standartmetodes precizitātes starprādītāji.";
4) LVS ISO 5725-4:2006 "Mērīšanas metožu precizitāte (rezultātu patiesums un konverģentums) – 4.daļa: Vienmetodes standartmērījumu patiesuma noteikšanas pamatmetodes.";
5) LVS ISO 5725-5:2006 "Mērīšanas metožu precizitāte (rezultātu patiesums un konverģentums) – 5.daļa: Alternatīvas metodes standarta mērīšanas metodēm precizitātes noteikšanai.";
6) LVS ISO 5725-6:2006 "Mērīšanas metožu un rezultātu pareizība (patiesums un precizitāte). 6.daļa: Pareizības vērtību lietošana praksē.
Veselības ministre Ingrīda Circene
3.pielikums
Ministru kabineta
2013.gada 2.jūlija
noteikumiem Nr.354
Kosmētikas līdzekļa mikrobioloģiskās tīrības kritēriji
|
Nr. p.k. |
Kosmētikas līdzekļa veids |
Mezofilo aerobo mikroorganismu daudzums (KVV/g vai ml) kosmētikas līdzeklī |
Stafilococcus aureus |
Pseudomonas aeruginosa |
Candida albicans |
|
1. |
Kosmētikas līdzekļi, kas paredzēti bērniem līdz trim gadiem, lietošanai acu tuvumā un uz gļotādām |
ne vairāk par 102 KVV 0,5 gramos (0,5 ml) kosmētikas līdzekļa |
0,5 gramos (0,5 ml) kosmētikas līdzekļa nedrīkst būt neviena KVV |
0,5 gramos (0,5 ml) kosmētikas līdzekļa nedrīkst būt neviena KVV |
0,5 gramos (0,5 ml) kosmētikas līdzekļa nedrīkst būt neviena KVV |
|
2. |
Pārējie kosmētikas līdzekļi |
ne vairāk par 103 KVV 0,1 gramā (0,1 ml) kosmētikas līdzekļa |
0,1 gramā (0,1 ml) kosmētikas līdzekļa nedrīkst būt neviena KVV |
0,1 gramā (0,1 ml) kosmētikas līdzekļa nedrīkst būt neviena KVV |
0,1 gramā (0,1 ml) kosmētikas līdzekļa nedrīkst būt neviena KVV |
Veselības ministre Ingrīda Circene
4.pielikums
Ministru kabineta
2013.gada 2.jūlija
noteikumiem Nr.354
Iesniegums brīvās tirdzniecības sertifikāta saņemšanai
Veselības inspekcijai
1. Informācija par iesniedzēju
|
Nosaukums |
|
|
Reģistrācijas numurs komercreģistrā |
|
|
Juridiskā adrese |
|
|
Kontaktinformācija |
|
|
(vārds, uzvārds, tālruņa numurs, e-pasts) |
2. Kosmētikas līdzekļa ražošanas vietas adrese
3. Informācija par atbildīgo personu
|
Atbildīgās personas nosaukums un adrese |
|
|
Adrese, kurā pieejama kosmētikas līdzekļa lieta |
4. Apliecinājums
Apliecinu, ka pielikumā, kas ir šī iesnieguma neatņemama sastāvdaļa, minētie kosmētikas līdzekļi atbilst prasībām, kas noteiktas Eiropas Parlamenta un Padomes regulā (EK) Nr.1223/2009 par kosmētikas līdzekļiem.
5. Lūdzu izsniegt brīvās tirdzniecības sertifikātu
|
kosmētikas līdzekļu eksportam uz |
|
|
(valsts nosaukums(-i)) |
|
latviešu valodā |
|
angļu valodā |
|
|
(eksemplāru skaits) |
(eksemplāru skaits) |
Datums ______________________________
|
Iesniedzējs |
|
|
(vārds, uzvārds, paraksts) |
Piezīme. Dokumenta rekvizītus "paraksts" un "datums" neaizpilda, ja elektroniskais dokuments ir sagatavots atbilstoši normatīvajiem aktiem par elektronisko dokumentu noformēšanu.
Pielikums
iesniegumam brīvās tirdzniecības
sertifikāta
saņemšanai
Kosmētikas līdzekļu saraksts
|
Nr. |
Kosmētikas līdzekļa nosaukums1 |
|
1. 2. ..
|
Piezīme. 1 Kosmētikas līdzekļa kategorija un nosaukums vai nosaukumi, kas ļauj to identificēt (ja nosaukums minēts vairākās valodās, norāda to nosaukumu, kurš nepieciešams sertifikātā).
Veselības ministre Ingrīda Circene
