Precizēts 03.06.2004., Latvijas
Vēstnesis Nr.89 (3037)
Ministru kabineta noteikumi
Nr.418
Rīgā 2004.gada 22.aprīlī (prot.
Nr.24, 55.§)
Grozījumi Ministru
kabineta 2000.gada 31.oktobra noteikumos Nr.381 “Zāļu
reģistrēšanas noteikumi”
Izdoti saskaņā
ar Farmācijas likuma 5.panta 3.punktu
Izdarīt Ministru kabineta
2000.gada 31.oktobra noteikumos Nr.381 “Zāļu reģistrēšanas
noteikumi” (Latvijas Vēstnesis, 2000, 391./393.nr.; 2003,
116.nr.) šādus grozījumus:
1. Izteikt 1.punktu šādā
redakcijā:
“1. Noteikumi nosaka rūpnieciski
ražoto zāļu (izņemot veterinārās zāles un veterinārfarmaceitiskos
produktus) reģistrēšanas kārtību.”
2. Papildināt noteikumus ar
1.1 punktu šādā redakcijā:
“1.1 Noteikumi
neattiecas uz:
1.1 1. zālēm, kas
izgatavotas saskaņā ar recepti (jebkura recepte, ko izraksta
speciālists, kura kvalifikācija atļauj to darīt) konkrētam
pacientam (formula magistralis);
1.1 2. zālēm, kas
izgatavotas aptiekā, atbilst farmakopejas monogrāfijām un ko
paredzēts izplatīt pacientiem, kurus apkalpo attiecīgā aptieka
(formula officinalis);
1.1 3. zālēm, kas
paredzētas pētījumiem, kā arī uz zāļu izpēti to izstrādes
procesā;
1.1 4. starpproduktiem,
ko licencēts zāļu ražotājs paredzējis turpmākai pārstrādei;
1.1 5. radionuklīdiem
(radioaktīviem izotopiem) slēgtu starojuma avotu veidā;
1.1 6. asinīm, cilvēka
izcelsmes asins plazmu un asins šūnām.”
3. Papildināt 2.punktu ar 2.3. un
2.4.apakšpunktu šādā redakcijā:
“2.3. tajā skaitā šo noteikumu
18.16.apakšpunktā minētos ģeneratorus un 36.7.apakšpunktā minētos
radiofarmaceitiskos produktus, radiofarmaceitiskos komplektus
(kitus) un citus radionuklīdus;
2.4. tajā skaitā minerālūdeņus, ja
tie atbilst zāļu definīcijai un Valsts zāļu aģentūra tos ir
atzinusi par zālēm.”
4. Aizstāt 3.1.apakšpunktā vārdus
“orāli (ieņemot caur muti)” ar vārdu “perorāli”.
5. Izteikt 8.punktu šādā
redakcijā:
“8. Valsts zāļu aģentūra zāļu
reģistrēšanas procedūru pabeidz 210 dienu laikā, ņemot vērā šo
noteikumu 136.punkta nosacījumus. Ja reģistrācijas pieprasījumā
ir norādīts, ka zāles reģistrē savstarpējā zāļu atzīšanas
procedūrā un reģistrācijas pieprasījumu par tām pašām zālēm
izskata cita Eiropas Savienības dalībvalsts vai Eiropas brīvās
tirdzniecības asociācijas valsts (EBTA valsts), kas ir
parakstījusi Eiropas Ekonomiskās zonas līgumu, kompetentā
institūcija (turpmāk — dalībvalsts), un reģistrācijas
pieprasītājs (pretendents reģistrācijas apliecības turēšanai)
informē Valsts zāļu aģentūru par atsauces dalībvalsti (zāļu
savstarpējā reģistrācijas atzīšanas procedūrā nozīmē to
dalībvalsti, kura zāles reģistrē sākotnēji), Valsts zāļu aģentūra
pieņem lēmumu par reģistrācijas pieprasījuma izskatīšanas
apturēšanu līdz dienai, kad saņemts atsauces dalībvalsts (kuru
par tādu ir atzinis reģistrācijas pieprasītājs) apstiprināts zāļu
analītiskās, farmakotoksikoloģiskās un klīniskās izpētes
rezultātu novērtējuma ziņojums (turpmāk — novērtējuma
ziņojums), un par pieņemto lēmumu informē atsauces dalībvalsti un
pretendentu.”
6. Papildināt noteikumus ar
8.1 punktu šādā redakcijā:
“8.1 Ja zāles Valsts
zāļu aģentūrā ir reģistrācijas procesā un reģistrācijas
pieprasītājs par atsauces dalībvalsti ir norādījis Latviju, bet
Valsts zāļu aģentūra ir saņēmusi informāciju no citas dalībvalsts
par to, ka tā ir pieņēmusi lēmumu apturēt attiecīgo zāļu
reģistrācijas pieprasījuma detalizētu izskatīšanu, lai sagaidītu
Valsts zāļu aģentūras apstiprinātu novērtējuma ziņojumu, Valsts
zāļu aģentūra pēc reģistrācijai iesniegto datu un dokumentu
pārbaudes un lēmuma pieņemšanas par zāļu reģistrāciju nosūta
novērtējuma ziņojuma kopiju attiecīgajai dalībvalstij.”
7. Izteikt 9.punktu šādā
redakcijā:
“9. Ja saskaņā ar šo noteikumu
18.11.apakšpunktu Valsts zāļu aģentūru informē par to, ka zāles,
uz kurām attiecas reģistrācijas pieprasījums, ir reģistrētas
dalībvalstīs, un pieprasījumā norādīts, ka pieprasījums attiecas
uz savstarpējo reģistrācijas atzīšanas procedūru, Valsts zāļu
aģentūra:
9.1. aptur reģistrācijas
pieprasījuma izskatīšanu un nosūta atsauces dalībvalstij lūgumu
atsūtīt tās apstiprināto novērtējuma ziņojumu;
9.2. 90 dienu laikā pēc
novērtējuma ziņojuma saņemšanas atzīst atsauces dalībvalsts zāļu
reģistrāciju un zāļu aprakstu, kuru tā ir apstiprinājusi, un
reģistrē zāles vai, ja tā uzskata, ka attiecīgo zāļu reģistrācija
(atļaušana) rada risku iedzīvotāju veselībai, piemēro šo
noteikumu I1 nodaļā minētās procedūras.”
8. Papildināt noteikumus ar
I1 nodaļu šādā redakcijā:
“I1
Savstarpējā zāļu reģistrācijas atzīšana
12.1 Zāļu reģistrācijas
apliecības (tirdzniecības atļaujas) īpašnieks (turpmāk —
reģistrācijas īpašnieks):
12.1 1. pirms
pieprasījuma iesniegšanas attiecīgajai dalībvalstij informē
atsauces dalībvalsts kompetento institūciju (Latvijā —
Valsts zāļu aģentūru) par to, ka pieprasījumu paredzēts iesniegt
citai dalībvalstij reģistrācijas atzīšanai, un paziņo par
jebkuriem reģistrācijas dokumentācijas papildinājumiem;
12.1 2. lūdz atsauces
dalībvalsts kompetento institūciju sagatavot vai, ja
nepieciešams, atjaunot novērtējuma ziņojumu par reģistrētajām
zālēm, kurām paredzēts uzsākt savstarpējo reģistrācijas atzīšanas
procedūru;
12.1 3. atbilstoši šo
noteikumu 15.punktā noteiktajām prasībām iesniedz dalībvalsts
kompetentajā institūcijā pieprasījumu reģistrācijas savstarpējai
atzīšanai, pievienojot šajos noteikumos noteiktos datus un
dokumentus, un apliecina, ka:
12.1 3.1. reģistrācijas
dokumentācija ir identiska atsauces dalībvalsts kompetentajā
institūcijā iesniegtajai informācijai, vai norāda visus tajā
izdarītos grozījumus;
12.1 3.2. saskaņā ar šo
noteikumu 36.punktā noteiktajām prasībām iesniegtais zāļu
apraksts ir identisks atsauces dalībvalstī apstiprinātajam zāļu
aprakstam;
12.1 3.3. visa
dokumentācija, kas ir iesniegta dalībvalstīs kā procedūras daļa,
ir identiska;
12.1 4. paziņo Eiropas
Zāļu novērtēšanas aģentūrai, kas izveidota ar Padomes 1993.gada
22.jūlija Regulu (EEK) Nr.2309/93, ar ko nosaka Eiropas Kopienas
procedūru tam, kā apstiprināt un pārraudzīt zāles, kuras
paredzētas izmantošanai cilvēkiem un veterinārijā, un ar ko
nodibina Eiropas Zāļu novērtēšanas aģentūru (turpmāk —
Padomes regula Nr.2309/93) (turpmāk — Eiropas Zāļu
novērtēšanas aģentūra), par iesaistītajām dalībvalstīm un
pieprasījuma iesniegšanas datumiem, kā arī nosūta tai citu
dalībvalstu izsniegtās reģistrācijas apliecību kopijas un norāda,
vai pieprasījumu pašlaik izskata kāda cita dalībvalsts;
12.1 5. ir tiesīgs
jautājumu iesniegt izskatīšanai Eiropas Zāļu novērtēšanas
aģentūras Zāļu komitejā (angliski — Committee for
Proprietary Medicinal Products) (turpmāk — Zāļu
komitejai), lai tā uzsāktu reģistrācijas jautājuma izskatīšanas
procedūru:
12.1 5.1. ja
pieprasījumi ir iesniegti vairākās dalībvalstīs un dalībvalstis
ir pieņēmušas atšķirīgus lēmumus par zāļu reģistrēšanu,
reģistrācijas apturēšanu (zāļu reģistrēšanas apliecības darbības
apturēšanu) vai reģistrācijas anulēšanu. Zāļu komitejai iesniedz
visu par attiecīgo jautājumu pieejamo informāciju, un iesniegtais
jautājums ir skaidri identificēts. Reģistrācijas īpašniekam ir
tiesības sniegt paskaidrojumu rakstiski vai mutiski;
12.1 5.2. ja ir skartas
Eiropas Kopienas intereses, pirms dalībvalsts ir pieņēmusi lēmumu
par zāļu reģistrāciju vai reģistrācijas apturēšanu, vai
reģistrācijas anulēšanu, vai citām izmaiņām reģistrācijas
apliecības nosacījumos (izmaiņas reģistrācijas dokumentācijā),
īpaši ņemot vērā informāciju, kas iegūta, uzraugot zāļu
lietošanas izraisītās blakusparādības (blaknes). Zāļu komitejai
iesniedz visu par attiecīgo jautājumu pieejamo informāciju, un
iesniegtais jautājums ir skaidri identificēts;
12.1 6. pēc šo
noteikumu 12.3 2.apakšpunktā minētās informācijas
saņemšanas iesniedz Zāļu komitejai tos datus, dokumentus un
informāciju, kura ir iesniegta dalībvalstij saskaņā ar šo
noteikumu 12.1 3.apakšpunktu;
12.1 7. ir tiesīgs 15
dienu laikā pēc Eiropas Zāļu novērtēšanas aģentūras atzinuma
saņemšanas rakstiski paziņot Eiropas Zāļu novērtēšanas aģentūrai
par nodomu apstrīdēt Eiropas Zāļu novērtēšanas aģentūras atzinumu
(apelācijas sūdzība) un 60 dienu laikā no atzinuma saņemšanas
dienas iesniegt Eiropas Zāļu novērtēšanas aģentūrā detalizētu
apelācijas pamatojumu;
12.1 8. pieprasījumu
par izmaiņām reģistrācijas apliecībā (reģistrācijas
dokumentācijā) iesniedz attiecīgajām dalībvalstīm, kurās zāles ir
reģistrētas, un izskata izmaiņas saskaņā ar Eiropas Komisijas
2003.gada 3.jūnija Regulu (EK) Nr.1084/2003 par izmaiņu
izskatīšanu dalībvalsts kompetentas iestādes izsniegtas cilvēkiem
paredzētu zāļu un veterināro zāļu tirdzniecības atļaujas
nosacījumos (turpmāk — Komisijas regula Nr.1084/2003).
12.2 Ja atsauces
dalībvalsts ir Latvija, Valsts zāļu aģentūra:
12.2 1. ir tiesīga
pieprasīt reģistrācijas īpašniekam pēc šo noteikumu
12.1 1.apakšpunktā minētās informācijas saņemšanas
iesniegt datus un dokumentus, lai pārbaudītu, vai reģistrācijas
dokumentācija ir identiska Valsts zāļu aģentūrā iesniegtajai
informācijai;
12.2 2. sagatavo vai
atjauno novērtējuma ziņojumu 90 dienu laikā no šo noteikumu
12.1 2.apakšpunktā minētā lūguma saņemšanas un nosūta
novērtējuma ziņojumu attiecīgai dalībvalstij pēc tam, kad tajā ir
iesniegts pieprasījums, kā arī sadarbojas ar attiecīgajām
dalībvalstīm, lai panāktu vienošanos par iesniegto
pieprasījumu.
12.3 Valsts zāļu
aģentūra:
12.3 1. atzīst atsauces
dalībvalsts izdoto zāļu reģistrācijas apliecību 90 dienu laikā
pēc attiecīgā pieprasījuma un novērtējuma ziņojuma saņemšanas, ja
nav pamata uzskatīt, ka attiecīgo zāļu reģistrācija var radīt
draudus sabiedrības veselībai. Par pieņemto lēmumu informē
atsauces dalībvalsti, iesaistītās dalībvalstis, Eiropas Zāļu
novērtēšanas aģentūru un reģistrācijas īpašnieku (pieprasījuma
iesniedzēju);
12.3 2. ja ir pamats
uzskatīt, ka attiecīgo zāļu reģistrācija var radīt draudus
sabiedrības veselībai, informē atsauces dalībvalsti, iesaistītās
dalībvalstis, Eiropas Zāļu novērtēšanas aģentūru un reģistrācijas
īpašnieku (pieprasījuma iesniedzēju), detalizēti izklāsta lēmuma
pamatojumu un identificē trūkumu novēršanai nepieciešamās
darbības. Valsts zāļu aģentūra sadarbojas ar attiecīgajām
dalībvalstīm, lai vienotos par rīcību, un dod iespēju
pieprasījuma iesniedzējam sniegt paskaidrojumus rakstiski vai
mutiski;
12.3 3. ir tiesīga
iesniegt jautājumu izskatīšanai Zāļu komitejā, lai tā uzsāktu
jautājuma izskatīšanas procedūru:
12.3 3.1. ja nav
panākta vienošanās šo noteikumu 12.3 1.apakšpunktā
minētajā termiņā par šo noteikumu 12.3 2.apakšpunktā
ietverto jautājumu. Zāļu komitejai un pieprasījuma iesniedzējam
iesniedz detalizētu jautājuma izklāstu, par kuru nav panākta
vienošanās, un norāda nesaskaņu iemeslus;
12.3 3.2. ja
pieprasījumi ir iesniegti vairākās dalībvalstīs un tās ir
pieņēmušas atšķirīgus lēmumus par zāļu reģistrēšanu,
reģistrācijas apturēšanu un reģistrācijas anulēšanu. Zāļu
komitejai iesniedzamo jautājumu skaidri definē un par to informē
reģistrācijas īpašnieku. Zāļu komitejai iesniedz visu par
attiecīgo jautājumu pieejamo informāciju;
12.3 3.3. ja ir skartas
Eiropas Kopienas intereses, pirms dalībvalsts ir pieņēmusi lēmumu
par zāļu reģistrāciju vai reģistrācijas apturēšanu, vai
reģistrācijas anulēšanu, vai citām izmaiņām reģistrācijas
apliecības nosacījumos (izmaiņas reģistrācijas dokumentācijā),
īpaši ņemot vērā informāciju, kas iegūta, uzraugot zāļu
lietošanas izraisītās blakusparādības (blaknes). Zāļu komitejai
iesniedz visu par attiecīgo jautājumu pieejamo informāciju, un
iesniegtais jautājums ir skaidri identificēts;
12.3 4. 28 dienu laikā
pēc tam, kad saņemts Eiropas Komisijas sagatavotais lēmumprojekts
par reģistrācijas pieprasījumu sakarā ar Eiropas Zāļu
novērtēšanas aģentūras atzinumu, var rakstiski iesniegt
Pastāvīgajā komitejā (angliski — Standing Committee)
apsvērumus par lēmumprojektu un pieprasīt tā apspriešanu
Pastāvīgajā komitejā, sniedzot detalizētu pamatojumu;
12.3 5. 30 dienu laikā
pēc Eiropas Komisijas galīgā lēmuma paziņošanas atbilstoši
lēmumam zāles reģistrē vai reģistrāciju anulē, vai izdara
izmaiņas reģistrācijas apliecības nosacījumos (reģistrācijas
dokumentācijā) un informē par to Eiropas Komisiju un Eiropas Zāļu
novērtēšanas aģentūru;
12.3 6. iesniedz
jautājumu izskatīšanai Eiropas Zāļu novērtēšanas aģentūrā, ja
uzskata, ka izmaiņas zāļu reģistrācijas apliecībā (izmaiņas
reģistrācijas dokumentācijā) vai reģistrācijas apturēšana, vai
reģistrācijas anulēšana ir nepieciešama sabiedrības veselības
aizsardzībai;
12.3 7. izņēmuma
gadījumā, ja iedzīvotāju veselības aizsardzībai būtiska ir
steidzama rīcība, neskarot šo noteikumu
12.3 3.3.apakšpunkta nosacījumus, var pieņemt
lēmumu par zāļu reģistrācijas apliecības darbības apturēšanu tās
teritorijā (lai novērstu zāļu izplatīšanu un lietošanu) līdz
galīgā lēmuma pieņemšanai. Par pieņemto lēmumu dalībvalsts
informē Eiropas Komisiju un citas dalībvalstis ne vēlāk kā
nākamajā darbdienā pēc lēmuma pieņemšanas.
12.4 Šajā nodaļā
noteiktās prasības nepiemēro homeopātiskajām zālēm, uz kurām
attiecina īpašus noteikumus toksikoloģiskajām un
farmakoloģiskajām pārbaudēm un klīniskajai izpētei saskaņā ar
attiecīgās dalībvalsts homeopātijas principiem un iezīmēm
atbilstoši šo noteikumu 29.1.apakšpunktam.”
9. Izteikt 13., 14. un 15.punktu
šādā redakcijā:
“13. Reģistrācijas un
pārreģistrācijas pieprasītājs:
13.1. nodrošina zāļu reģistrācijai
un pārreģistrācijai nepieciešamās informācijas un datu
sagatavošanu un iesniegšanu saskaņā ar šajos noteikumos
noteiktajām prasībām;
13.2. sastāda un iesniedz Valsts
zāļu aģentūrā zāļu reģistrācijas un/vai pārreģistrācijas
pieprasījumu un tam pievieno datus, dokumentus un informāciju,
kas sastādīta atbilstoši Farmācijas likuma 28.1 pantā
minētajiem Eiropas Komisijas ieteikumiem par dokumentāciju un
prasībām zāļu kvalitātei, drošumam un efektivitātei, ko Eiropas
Komisija publicējusi Eiropas Kopienas Zāļu tiesiskā regulējuma
sējumos;
13.3. nodrošina, ka šo noteikumu
18.7., 18.8. un 19.2.apakšpunktā minētos dokumentus un datus
pirms iesniegšanas Valsts zāļu aģentūrā sagatavo un paraksta
eksperti atbilstoši viņu kvalifikācijai un pienākumiem saskaņā ar
šo noteikumu 14.punktu. Ekspertīzes izmaksas sedz reģistrācijas
pieprasītājs.
14. Ekspertiem atbilstoši viņu
kvalifikācijai ir šādi pienākumi:
14.1. izpildīt uzdevumus, kas
ietilpst to pārstāvētajās attiecīgajās nozarēs (analītiskā
ķīmija, farmakoloģija un līdzīgas eksperimentālās zinātnes,
klīniskā izpēte), un objektīvi aprakstīt (kvalitatīvi un
kvantitatīvi) iegūtos rezultātus;
14.2. aprakstīt savus novērojumus
atbilstoši šo noteikumu 2.pielikumā “Analīzes,
farmakotoksikoloģijas un klīniskie standarti un protokoli
attiecībā uz zāļu testēšanu” noteiktajām prasībām, īpaši norādot
datus:
14.2.1. analītiskās ķīmijas
speciālistiem — par zāļu atbilstību norādītajam sastāvam,
sniedzot arī pamatojumu ražotāja izmantotajām kontroles
metodēm;
14.2.2. farmakologiem vai
speciālistiem ar līdzīgu pieredzi — par zāļu toksicitāti un
novērotajām farmakoloģiskajām īpašībām;
14.2.3. klīniskās izpētes
speciālistiem (ārstiem) — par zāļu klīniskās izpētes
rezultātiem (tādu zāļu iedarbību uz cilvēkiem, kas atbilst
reģistrācijas pieprasītāja sniegtajiem datiem saskaņā ar šo
noteikumu 18., 19. un 21.punktu, tajā skaitā par ārsta ieteikto
devu panesamību), kā arī par visām kontrindikācijām un
blakusprādībām;
14.3. ja nepieciešams, izvērtē
zinātniskās literatūras izmantošanu šo noteikumu 19.2.apakšpunktā
minētajā gadījumā.
15. Reģistrācijas īpašniekam
(persona, kura ir atbildīga par zāļu izplatīšanu) ir šādi
pienākumi:
15.1. nodrošināt reģistrēto zāļu
atbilstību reģistrācijas apliecības nosacījumiem (reģistrācijas
dokumentācijai);
15.2. iesniegt to dalībvalstu
kompetentajām institūcijām, kurās zāles ir reģistrētas
(Latvijā — Valsts zāļu aģentūrai), informāciju un datus par
izmaiņām reģistrācijas dokumentācijā (tajā skaitā reģistrācijas
apliecības paplašināšanai ), kā arī veikt steidzamus ar drošību
saistītus ierobežojumus (veikt pagaidu izmaiņas zāļu
aprakstā — terapeitiskajās indikācijās, zāļu devās,
kontrindikācijās, brīdinājumos un zāļu izdalīšanās periodā) un
nodrošināt procedūras saskaņā ar:
15.2.1. Komisijas regulu
Nr.1084/2003 zālēm, kuras ir reģistrētas saskaņā ar nacionālo
reģistrācijas procedūru un kuru reģistrācija ir pakļauta
savstarpējai zāļu reģistrācijas atzīšanas procedūrai (attiecas
arī uz zālēm, kuras ir atļāvusi Zāļu komiteja līdz 1995.gada
1.janvārim);
15.2.2. šajos noteikumos
noteiktajām prasībām zālēm, kuras ir reģistrētas saskaņā ar
nacionālo reģistrācijas procedūru (nav pakļautas zāļu
savstarpējai reģistrācijas atzīšanas procedūrai);
15.2.3. Eiropas Komisijas
2003.gada 3.jūnija Regulu (EK) Nr.1085/2003 par izmaiņu
izskatīšanu cilvēkiem paredzētu zāļu un veterināro zāļu
tirdzniecības atļaujas nosacījumos, uz ko attiecas regula, ar ko
nosaka Kopienas procedūru tam, kā apstiprināt un pārraudzīt
zāles, kuras paredzētas izmantošanai cilvēkiem un veterinārijā,
un ar ko nodibina Eiropas Zāļu novērtēšanas aģentūru
(turpmāk — Komisijas regula Nr.1085/2003) zālēm, kuras ir
reģistrētas saskaņā ar Padomes regulu Nr.2309/93;
15.3. sastādīt un iesniegt Valsts
zāļu aģentūrā pieprasījumu izmaiņu apstiprināšanai (arī
reģistrācijas apliecības paplašināšanai). Pieprasījumam pievienot
datus, dokumentus un informāciju, kas ir sastādīta atbilstoši šo
noteikumu 13.2.apakšpunktā minētajiem Eiropas Komisijas
ieteikumiem;
15.4. šo noteikumu 18.4. un
18.7.apakšpunktā norādīto informāciju par ražošanas un kontroles
metodēm sagatavot atbilstoši zinātnes un tehnikas attīstībai un
veikt izmaiņas, kas nepieciešamas, lai zāles ražotu un pārbaudītu
ar vispārpieņemtiem zinātniskiem paņēmieniem, kā arī nodrošināt,
ka minētās izmaiņas ir apstiprinājusi attiecīgās dalībvalsts
kompetentā iestāde. Ja nepieciešams, iesniegt apliecinājumus par
zāļu un/vai to sastāvdaļu (vielu) kontroli un ražošanas procesa
starpstadiju kontroli saskaņā ar šo noteikumu 18.7.apakšpunktā
paredzētajām metodēm. Nodrošināt, ka imunoloģisko preparātu
ražotāji pēc Valsts zāļu aģentūras pieprasījuma iesniedz Valsts
zāļu aģentūrā sērijas izlaides sertifikātu (kontroles ziņojumu)
kopijas, ko parakstījusi kvalificētā persona;
15.5. atkārtoti izvērtēt saskaņā
ar nacionālo reģistrācijas procedūru reģistrēto zāļu ieguvumu un
riska attiecību, pēc reģistrācijas apliecības saņemšanas
iesniedzot Valsts zāļu aģentūrā jaunāko informāciju, kas nav
iekļauta sākotnējā reģistrācijas pieprasījumā, un informāciju par
zāļu blakusparādību pārraudzību atbilstoši normatīvajiem aktiem
par zāļu lietošanas izraisīto blakusparādību uzraudzību. Ievērot
Farmācijas likuma 28.1 pantā minētajos Eiropas
Komisijas ieteikumos (vadlīnijās), ko Eiropas Komisija ir
publicējusi Eiropas Kopienas Zāļu tiesiskā regulējuma 9.sējumā,
iekļautās prasības;
15.6. informēt tās dalībvalstis,
kurās attiecīgās zāles izplata (Latvijā — Valsts zāļu
aģentūru):
15.6.1. par jebkuru zāļu
aizliegšanu, kā arī lietošanas ierobežojumu, ko pieņēmušas
attiecīgās dalībvalsts kompetentās institūcijas;
15.6.2. par jebkuru darbību, lai
zāles izņemtu no apgrozības un apturētu zāļu izplatīšanu, un
norāda iemeslu;
15.6.3. par iespējamām izmaiņām
reģistrācijas dokumentācijas datos un/vai dokumentos;
15.7. parakstīt ar zāļu ražotāju
līgumu, kas garantē, ka ražošanas operācijas atbilst normatīvajos
aktos par zāļu ražošanu un kontroli noteiktajām prasībām un
reģistrācijas dokumentācijā norādītajiem ražošanas apstākļiem, ja
reģistrācijas apliecības īpašnieks nav zāļu ražotājs;
15.8. nodrošināt zinātniska
dienesta pakalpojumu esību attiecībā uz attiecīgo zāļu
zinātniskās informācijas sastādīšanu, kā arī tās atjaunošanu;
15.9. uzturēt dokumentāciju un
datus par attiecīgajām zālēm un nodot arhīvam glabāšanā saskaņā
ar normatīvajos aktos par dokumentu glabāšanu noteiktajām
prasībām.”
10. Izteikt 18.punkta ievaddaļu
šādā redakcijā:
“18. Lai reģistrētu zāles (izņemot
šo noteikumu 22.punktā minēto gadījumu), reģistrācijas
pieprasītājs Valsts zāļu aģentūrā iesniedz pieprasījumu.
Pieprasījumā ietver datus un dokumentus un saskaņā ar šo
noteikumu prasībām norāda šādu informāciju:”
11. Svītrot 18.4.apakšpunktā vārdu
“īsu”.
12. Papildināt 18.9.apakšpunktu ar
otro teikumu šādā redakcijā:
“Marķējums un lietošanas
instrukcijas atbilst Farmācijas likuma 28.1 pantā minētajiem
Eiropas Komisijas ieteikumiem (vadlīnijām) par dokumentāciju un
prasībām par zāļu kvalitāti, drošumu un efektivitāti, ko Eiropas
Komisija ir publicējusi Eiropas Kopienas Zāļu tiesiskā regulējuma
sējumā iekļautajām marķējuma un lietošanas instrukcijas
salasāmības prasībām, un tajā ir norādītas palīgvielas atbilstoši
minētajiem ieteikumiem.”
13. Izteikt 19.2.apakšpunktu šādā
redakcijā:
“19.2. zāļu sastāvdaļu(-as)
lietošana medicīnā ir plaša (vispāratzīta, labi ieviesusies), ar
atzītu iedarbību un pieņemamu drošības līmeni, norādot atsauces
uz zinātnisko literatūru;”.
14. Aizstāt 19.3. un
19.4.apakšpunktā vārdus “šo noteikumu 20.punktā minētajām zālēm,
kuras iegūtas, izmantojot attīstītās tehnoloģijas metodes” ar
vārdiem “augstās tehnoloģijas zālēm, kuras Zāļu komiteja līdz
1995.gada 1.janvārim ir atļāvusi dalībvalstīs”.
15. Izteikt 24.3.apakšpunktu šādā
redakcijā:
“24.3. klīniskās izpētes ekspertu
ziņojumu un citus paziņojumus un sertifikātus, ja tādi ir
vajadzīgi;”.
16. Papildināt noteikumus ar
24.5.apakšpunktu šādā redakcijā:
“24.5. attiecīgās dalībvalsts
izsniegtu apliecinājumu par atbilstību labai ražošanas praksei
(ne vecāku par trim gadiem).”
17. Papildināt noteikumus ar
26.1 punktu šādā redakcijā:
“26.1 Datus un
dokumentus, kurus pievieno reģistrācijas pieprasījumam
(reģistrācijas dokumentācija), Valsts zāļu aģentūrā iesniedz
saskaņā ar šo noteikumu 2.pielikumā noteiktajām prasībām, kā arī
ņemot vērā Farmācijas likuma 28.1 pantā minētos Eiropas Komisijas
ieteikumus (vadlīnijas) par dokumentāciju, ko Eiropas Komisija
publicējusi Eiropas Kopienas Zāļu tiesiskā regulējuma 2.sējuma B
daļā. Reģistrācijas pieprasījuma iesniedzēji reģistrācijas
dokumentācijas sagatavošanā (komplektēšanā) (tajā skaitā zāļu
pārreģistrācijas un izmaiņu reģistrācijas dokumentācijā) ņem vērā
zinātniskās pamatnostādnes, kas attiecas uz cilvēkiem paredzētu
zāļu kvalitāti, drošību un efektivitāti, kā to pieņēmusi Zāļu
komiteja un publicējusi Eiropas Zāļu novērtēšanas aģentūra, kā
arī ņem vērā pārējās Eiropas Kopienas farmaceitiskās
pamatnostādnes, ko Eiropas Komisija ir publicējusi Eiropas
Kopienas Zāļu tiesiskā regulējuma sējumos.”
18. Papildināt noteikumus ar
27.1 punktu šādā redakcijā:
“27.1 Reģistrācijas dokumentācijas
datus un dokumentus iesniedz piecos moduļos (kopējs tehniskais
dokuments, turpmāk — KTD) saskaņā ar šo noteikumu
2.pielikumā noteiktajām prasībām un ievēro to formu, saturu un
numurēšanas sistēmu, kas izklāstīta šo noteikumu
26.11.apakšpunktā minētā 2.sējuma B daļā. KTD noformējums
attiecas uz visiem reģistrācijas pieprasījumiem. Reģistrācijas
dokumentācijas kvalitātes daļā (ķīmiskā, farmaceitiskā un
bioloģiskā dokumentācija) norāda Eiropas Farmakopejas
monogrāfijas, tajā skaitā vispārīgās monogrāfijas un vispārīgās
sadaļas. Reģistrācijas dokumentācijā norādītais ražošanas process
atbilst normatīvajiem aktiem par zāļu ražošanu un kontroli un
Farmācijas likuma 51.1 pantā minētiem zāļu labas
ražošanas prakses principiem un pamatnostādnēm, ko Eiropas
Komisija publicējusi Eiropas Kopienas Zāļu tiesiskā regulējuma
4.sējumā.”
19. Izteikt 29.punktu šādā
redakcijā:
“29. Reģistrācijas pieprasījumā
(reģistrācijas dokumentācijā) iekļauj visu informāciju, kas
attiecas uz konkrēto zāļu novērtēšanu, neatkarīgi no tā, vai tā
ir labvēlīga vai nelabvēlīga, īpaši uzsverot informāciju par
nepabeigtiem un/vai pārtrauktiem farmakotoksikoloģiskiem un/vai
klīniskiem testiem vai izpēti, kā arī norādot pabeigtu klīnisko
izpēti par terapeitiskajām indikācijām, uz kurām neattiecas zāļu
reģistrācijas pieprasījums. Homeopātiskajām zālēm:
29.1. kuras neattiecas uz šo
noteikumu 4.punktā minētājām zālēm, toksikoloģiskās un
farmakoloģiskās pārbaudes un klīniskās izpētes prasības īsteno,
ņemot vērā Latvijā praktizētās homeopātijas principus un
iezīmes;
29.2. kuras ir reģistrētas līdz
1993.gada 31.decembrim, neatkarīgi no tā, vai pēc minētā datuma
zāles ir pārreģistrētas saskaņā ar attiecīgās dalībvalsts tiesību
aktiem, Valsts zāļu aģentūra minēto zāļu reģistrācijā,
pārreģistrācijā vai izmaiņu apstiprināšanā ņem vērā reģistrāciju,
ko iepriekš piešķīrusi cita dalībvalsts. Valsts zāļu aģentūra ir
tiesīga nepiemērot terapeitiskās iedarbības pierādīšanas
prasību.”
20. Papildināt 30.punktu ar trešo
un ceturto teikumu šādā redakcijā:
“Zāļu klīniskā izpēte atbilst
normatīvajiem aktiem par zāļu klīnisko izpēti, tajā skaitā, ja
klīniskā izpēte notikusi trešajās valstīs (valstis, kuras nav
Eiropas Savienības dalībvalstis vai Eiropas brīvās tirdzniecības
asociācijas valstis (EBTA valstis), kas parakstījušas Eiropas
Ekonomiskās zonas līgumu). Neklīniskos (farmakotoksikoloģiskos)
pētījumus veic saskaņā ar labas laboratoriju prakses
prasībām.”
21. Svītrot 32.punktu.
22. Izteikt IX nodaļu šādā
redakcijā:
“IX. Izmaiņas
reģistrācijas dokumentācijā, par kurām iesniedz papildu
iesniegumu reģistrācijas apliecības paplašināšanai
111. Saskaņā ar nacionālo
reģistrācijas procedūru reģistrētām zālēm, kuras nepakļauj
savstarpējai reģistrācijas atzīšanai, izmaiņas reģistrācijas
dokumentācijā, par kurām iesniedz papildu iesniegumu
reģistrācijas apliecības paplašināšanai, ir noteiktas šo
noteikumu 3.pielikumā. Zāļu nosaukums reģistrācijas apliecības
paplašināšanas iesniegumā ir tāds pats kā zāļu reģistrācijas
apliecībā.
112. Valsts zāļu aģentūra izvērtē
iesniegto pieprasījumu un tam pievienotos datus un dokumentus un
pieņem lēmumu par izmaiņu apstiprināšanu zāļu reģistrācijas
apliecības paplašināšanai vai izmaiņu apstiprināšanu saskaņā ar
šo noteikumu X vai XI nodaļas prasībām, vai pieņem lēmumu atteikt
izmaiņu apstiprināšanu.”
23. Izteikt X nodaļu šādā
redakcijā:
“X. Nelielas
izmaiņas reģistrācijas dokumentācijā
113. Saskaņā ar nacionālo
reģistrācijas procedūru reģistrētām zālēm, kuras nepakļauj
savstarpējai reģistrācijas atzīšanai, nelielas IA tipa un IB tipa
izmaiņas reģistrācijas dokumentācijā ir noteiktas šo noteikumu
4.pielikumā.
114. Reģistrācijas īpašnieks
iesniedz Valsts zāļu aģentūrā pieprasījumu par izmaiņu
apstiprināšanu. Pieprasījumam pievieno:
114.1. grozītos datus, dokumentus
un citu informāciju;
114.2. dokumentu, kas apliecina
maksājumu par novērtēšanu saskaņā ar šo noteikumu 6.punktu.
115. Pieprasījumā norāda tikai
vienas I tipa izmaiņas. Ja izdarāmas vairākas I tipa izmaiņas,
par katru iesniedz atsevišķu pieprasījumu. Katrā pieprasījumā ir
atsauce uz pārējiem pieprasījumiem, ja tādi ir.
116. Ja no vienām IA tipa izmaiņām
izriet vairākas IA tipa izmaiņas, par katru izrietošo izmaiņu
reģistrācijas īpašnieks iesniedz atsevišķu pieprasījumu. Ja no
vienām IB tipa izmaiņām izriet IA tipa vai IB tipa izmaiņas, par
katru izrietošo izmaiņu reģistrācijas īpašnieks iesniedz
atsevišķu pieprasījumu par IB tipa izmaiņām. Pieprasījumā norāda
saistību starp visām izrietošajām I tipa izmaiņām.
117. Ja no I tipa izmaiņām izriet,
ka zāļu apraksts un/vai marķējums un/vai lietošanas instrukcija
ir grozāma, to uzskata par izmaiņu daļu.
118. Ja pieprasījums par izmaiņu
apstiprināšanu atbilst šo noteikumu 114., 115., 116., 117. un
118.punktā noteiktajiem nosacījumiem, Valsts zāļu aģentūra 14
darbdienu laikā no pieprasījuma saņemšanas dienas atzīst, ka
pieprasījums ir derīgs, un attiecīgi informē reģistrācijas
apliecības īpašnieku. Valsts zāļu aģentūra izskata pieprasījumu
un:
118.1. pēc pieprasījuma un tam
pievienoto datu, dokumentu un citas informācijas novērtēšanas
pieņem lēmumu par nelielu IA tipa izmaiņu apstiprināšanu vai
neapstiprināšanu un paziņo par to reģistrācijas īpašniekam;
118.2. ja pēc pieprasījuma un tam
pievienoto datu, dokumentu un citas informācijas novērtēšanas
Valsts zāļu aģentūra uzskata, ka pieprasījums par nelielām IB
tipa izmaiņām nav pieņemams, tā 30 dienu laikā pēc apliecinājuma
par derīga pieprasījuma saņemšanu, pamatojot savu atzinumu,
informē reģistrācijas īpašnieku, kurš ir iesniedzis attiecīgo
pieprasījumu. 30 dienu laikā pēc Valsts zāļu aģentūras atzinuma
saņemšanas reģistrācijas īpašniekam ir tiesības grozīt
pieprasījumu, ievērojot Valsts zāļu aģentūras atzinumā minēto
pamatojumu. Ja reģistrācijas īpašnieks pieprasījumu negroza,
pieprasījumu uzskata par noraidītu, un Valsts zāļu aģentūra
pieņemto lēmumu par izmaiņu neapstiprināšanu paziņo reģistrācijas
īpašniekam. Ja Valsts zāļu aģentūra nav nosūtījusi reģistrācijas
īpašniekam lēmumu par izmaiņu neapstiprināšanu, uzskata, ka
Valsts zaļu aģentūra izmaiņas ir apstiprinājusi.
119. Ja izmaiņas ir saistītas ar
reģistrācijas īpašnieka nosaukuma un/vai adreses maiņu un
reģistrācijas īpašnieks nav tā pati persona, esošais
reģistrācijas īpašnieks iesniedz Valsts zāļu aģentūrā
pieprasījumu. Pieprasījumam pievieno šādus dokumentus un norāda
šādu informāciju:
119.1. dokumentu, kas apliecina
īpašnieka maiņu;
119.2. zāļu nosaukumu,
reģistrācijas apliecības numuru un zāļu reģistrācijas datumu;
119.3. esošo reģistrācijas
īpašnieka nosaukumu un adresi, kā arī tās personas nosaukumu un
adresi, kurai paredzēts nodot reģistrācijas apliecību;
119.4. dokumentu, kas apliecina
pilnīgas un nodošanas brīdī atjaunotas reģistrācijas
dokumentācijas vai tās kopijas pieejamību un nodošanu personai,
kurai nodod reģistrācijas apliecību;
119.5. datumu, kad persona, kurai
paredzēts nodot reģistrācijas apliecību, varēs pārņemt no
iepriekšējā reģistrācijas īpašnieka visus zāļu reģistrācijas
īpašnieka pienākumus;
119.6. dokumentu, kas apliecina,
ka persona, kurai nodod reģistrācijas apliecību, spēj nodrošināt
reģistrācijas īpašnieka pienākumus atbilstoši prasībām, kas
noteiktas normatīvajos aktos par zāļu ražošanu un kontroli, zāļu
ievešanu, izvešanu un izplatīšanu, zāļu reklāmu, blakusparādībām
un zāļu klīnisko izpēti. Dokumentā norāda:
119.6.1. personu, kura atbildīga
par darbībām saistībā ar zāļu blakusparādību pārraudzības
sistēmu, tās adresi, tālruņa un faksa numuru. Pievieno īsu
darbības un pieredzes aprakstu;
119.6.2. dienestu, kurš atbildīgs
par zāļu reklamēšanu un izplatīšanu, kā arī attiecīgā dienesta
adresi, tālruņa un faksa numuru;
119.7. zāļu apraksta, lietošanas
instrukcijas un marķējuma projektu;
119.8. dokumentu, kas apliecina
maksājumu par novērtēšanu saskaņā ar šo noteikumu 6.punktu.
120. Valsts zāļu aģentūra
atbilstoši šo noteikumu 119.punktā minētajiem nosacījumiem
izmaiņas izskata kā IA tipa izmaiņas. Datumu, kad reģistrācijas
apliecība tiks nodota jaunajam reģistrācijas apliecības
īpašniekam, nosaka Valsts zāļu aģentūra, un tas tiek ierakstīts
līgumā starp esošo reģistrācijas īpašnieku un jauno reģistrācijas
īpašnieku. Reģistrācijas apliecības derīguma termiņš
nemainās.”
24. Izteikt XI nodaļu šādā
redakcijā:
“XI. Nozīmīgas
izmaiņas reģistrācijas dokumentācijā
121. Nozīmīgas II tipa izmaiņas ir
izmaiņas, kuras neatbilst šo noteikumu 3.pielikumā minētajām
izmaiņām vai izmaiņām, ko nosaka reģistrācijas apliecības satura
paplašināšana saskaņā ar šo noteikumu 4.pielikumu.
122. Reģistrācijas īpašnieks
iesniedz Valsts zāļu aģentūrā pieprasījumu nozīmīgu II tipa
izmaiņu apstiprināšanai. Pieprasījumam pievieno:
122.1. šo noteikumu 13., 14. un
15.punktā un III nodaļā minētos datus un pamatojošos dokumentus,
ekspertu ziņojumus, pārskatu un secinājumu pielikumus, kuri
aktualizēti atbilstoši izmaiņām reģistrācijas dokumentācijā, un
datus, kas pamato izmaiņas, par kurām iesniegts pieprasījums;
122.2. dokumentu, kas apliecina
maksājumu par novērtēšanu saskaņā ar šo noteikumu 6.punktu.
123. Pieprasījums attiecas tikai
uz vienu II tipa izmaiņu. Ja vienām reģistrētām zālēm jāveic
vairākas II tipa izmaiņas, par katru II tipa izmaiņu iesniedz
atsevišķu pieprasījumu. Katrā šādā pieprasījumā ir atsauce uz
pārējiem pieprasījumiem. Ja no vienām II tipa izmaiņām izriet
citas izmaiņas, par visām šādām izmaiņām var iesniegt vienu
pieprasījumu, un tajā norāda saistību starp visām izrietošajām
izmaiņām. Ja no izmaiņām izriet, ka zāļu apraksts un/vai
marķējums, un/vai lietošanas instrukcija ir grozāma, to uzskata
par izmaiņu daļu.
124. Ja pieprasījums atbilst šo
noteikumu 122. un 123.punktā noteiktajām prasībām, Valsts zāļu
aģentūra paziņo reģistrācijas īpašniekam, kad tiks uzsākta
pieprasījuma izskatīšana. 60 dienu laikā no pieprasījuma
izskatīšanas procedūras uzsākšanas Valsts zāļu aģentūra sagatavo
novērtējuma ziņojumu un lēmumprojektu. Minēto termiņu var
samazināt, ņemot vērā jautājuma steidzamību, jo īpaši saistībā ar
drošības jautājumiem.
125. Ja izmaiņas saistītas ar
terapeitisko indikāciju mainīšanu vai jaunu indikāciju
papildināšanu, minēto periodu var pagarināt līdz 90 dienām.
126. Valsts zāļu aģentūra ir
tiesīga lūgt reģistrācijas īpašnieku sniegt papildu informāciju.
Minētajā gadījumā procedūru aptur līdz laikam, kad reģistrācijas
īpašnieks iesniedz Valsts zāļu aģentūrā papildus pieprasīto
informāciju. Valsts zāļu aģentūra pieņem lēmumu par izmaiņu
apstiprināšanu vai neapstiprināšanu normatīvajos aktos noteiktajā
kārtībā, un par to paziņo reģistrācijas īpašniekam.
127. Atbilstoši izmaiņām cilvēkiem
paredzēto pretgripas vakcīnu reģistrācijas apliecības nosacījumos
(reģistrācijas dokumentācijā), piemēro šādas procedūras:
127.1. Valsts zāļu aģentūra 30
dienu laikā pēc procedūras uzsākšanas, pamatojoties uz kvalitāti
apliecinošiem dokumentiem, kas minēti šo noteikumu 2.pielikuma
3.modulī, sagatavo novērtējuma ziņojumu un lēmumprojektu un
nosūta reģistrācijas īpašniekam;
127.2. reģistrācijas īpašnieks 12
dienu laikā iesniedz Valsts zāļu aģentūrā klīniskos datus un, ja
nepieciešams, zāļu stabilitātes datus;
127.3. Valsts zāļu aģentūra
septiņu dienu laikā pēc datu saņemšanas novērtē šos datus un
sagatavo galīgo lēmumprojektu.
128. Sakarā ar cilvēka gripas
vīrusa pandēmiju (attiecas arī uz citām pandēmiskām cilvēka
slimībām), kuru par tādu atzinusi Pasaules Veselības organizācija
vai Eiropas Kopiena, Valsts zāļu aģentūrai izņēmuma kārtā ir
tiesības atzīt izmaiņas cilvēkiem paredzētās pretgripas vakcīnas
reģistrācijas apliecībā (reģistrācijas dokumentācijā) laikposmā
no pieprasījuma saņemšanas līdz šo noteikumu 127.punktā noteiktās
procedūras beigām, ja reģistrācijas īpašnieks iesniedz pilnīgus
datus par klīnisko drošību un efektivitāti.
129. Steidzamus ar drošību
saistītus ierobežojumus:
129.1. īsteno reģistrācijas
īpašnieks, ja rodas risks sabiedrības veselībai. Reģistrācijas
īpašnieks tajā paša dienā par to informē Valsts zāļu aģentūru. Ja
Valsts zāļu aģentūra 24 stundu laikā pēc minētās informācijas
saņemšanas nav izteikusi iebildumus, steidzamos ar drošību
saistītos ierobežojumus uzskata par atzītiem;
129.2. nosaka Valsts zāļu
aģentūra. Reģistrācijas īpašnieks iesniedz Valsts zāļu aģentūrā
pieprasījumu par izmaiņu apstiprināšanu, ņemot vērā Valsts zāļu
aģentūras noteiktos ierobežojumus.
130. Šo noteikumu 129.punktā
minētajā gadījumā steidzamos ar drošību saistītos ierobežojumus
reģistrācijas īpašnieks īsteno termiņā, par ko vienojas ar Valsts
zāļu aģentūru. Pieprasījumu par izmaiņām reģistrācijas
dokumentācijā atbilstoši steidzamajiem ar drošību saistītajiem
ierobežojumiem reģistrācijas īpašnieks iesniedz Valsts zāļu
aģentūrā ne vēlāk kā 15 dienu laikā pēc steidzamo ar drošību
saistīto ierobežojumu uzsākšanas.”
25. Papildināt noteikumus ar
135.4.apakšpunktu šādā redakcijā:
“135.4. ir tiesības ārkārtas
apstākļos (pēc apspriešanās ar reģistrācijas pieprasītāju)
izsniegt zāļu reģistrācijas apliecību, attiecinot uz to konkrētus
pienākumus, tajā skaitā pētījumu turpināšanu pēc reģistrācijas
apliecības saņemšanas un informēšanu par zāļu blakusparādībām.
Minētos lēmumus ir tiesības pieņemt objektīvu un pārbaudāmu
iemeslu dēļ, un to pamatā jābūt vienam no šo noteikumu
2.pielikuma 52.punktā minētajiem cēloņiem.”
26. Izteikt 136. un 137.punktu
šādā redakcijā:
“136. Lai izskatītu reģistrācijas
pieprasījumu, Valsts zāļu aģentūra, ja nepieciešams, ir tiesīga
lūgt reģistrācijas pieprasītājam iesniegt papildu datus
atbilstoši šo noteikumu 18., 19. un 21.punktā minētajām prasībām.
Šajā gadījumā šo noteikumu 8.punktā noteikto termiņa ierobežojumu
aptur līdz pieprasītās papildu informācijas saņemšanai. Termiņa
ierobežojumu aptur arī par laiku, kāds atvēlēts mutiska vai
rakstiska paskaidrojuma sniegšanai.
137. Valsts zāļu aģentūra,
izsniedzot reģistrācijas apliecību, informē zāļu reģistrācijas
īpašnieku par tās apstiprināto zāļu aprakstu un nodrošina zāļu
aprakstā sniegtās informācijas atbilstību reģistrācijas
dokumentācijai. Ja zāles ir reģistrētas zāļu savstarpējā
reģistrācijas atzīšanas procedūrā, Valsts zāļu aģentūra nosūta
Eiropas Zāļu novērtēšanas aģentūrai zāļu reģistrācijas apliecības
kopiju un zāļu aprakstu.”
27. Svītrot 138.punktā vārdus “un
komentārus”.
28. Papildināt 140.2.apakšpunktu
aiz vārda “noteikto” ar vārdiem “ja noskaidro, ka ar šīm zālēm
nevar iegūt ārstnieciskus rezultātus”.
29. Papildināt noteikumus ar
140.7.apakšpunktu šādā redakcijā:
“140.7. zāles ir iekļautas Eiropas
Kopienā aizliegto zāļu sarakstā.”
30. Papildināt 141.2.apakšpunktu
aiz vārda “punktu” ar vārdiem “ja noskaidro, ka ar šīm zālēm
nevar iegūt ārstnieciskus rezultātus”.
31. Papildināt noteikumus ar
141.1 punktu šādā redakcijā:
“141.1 Valsts zāļu
aģentūrai ir tiesības pieņemt lēmumu par steidzamu zāļu
reģistrācijas apturēšanu saskaņā ar zāļu blakusparādību
uzraudzības datu novērtējuma rezultātu, ja tā informē Eiropas
Zāļu novērtēšanas aģentūru, Eiropas Komisiju un pārējās
dalībvalstis ne vēlāk kā nākamajā darbdienā.”
32. Izteikt 143.punktu šādā
redakcijā:
“143. Valsts zāļu aģentūra,
pamatojoties uz zāļu reģistrēšanas komisijas ieteikumu, pieņem
lēmumu par zāļu:
143.1. reģistrāciju vai
reģistrācijas atteikumu;
143.2. pārreģistrāciju vai
pārreģistrācijas atteikumu;
143.3. reģistrācijas atteikšanas
atcelšanu, ja ir novērsti apstākļi, kuri bija par pamatu
attiecīgā lēmuma pieņemšanai;
143.4. reģistrācijas apturēšanu
vai, ja ir novērsti apstākļi, kuri bija par pamatu attiecīgā
lēmuma pieņemšanai, — reģistrācijas apturēšanas
atcelšanu;
143.5. reģistrācijas anulēšanu
vai, ja ir novērsti apstākļi, kuri bija par pamatu attiecīgā
lēmuma pieņemšanai, — reģistrācijas anulēšanas
atcelšanu;
143.6. reģistrācijas
dokumentācijas izmaiņu apstiprināšanu vai atteikumu apstiprināt
izmaiņas.”
33. Papildināt noteikumus ar
143.1 un 143.2 punktu šādā redakcijā:
“143.1 Ja pieņem lēmumu
par zāļu reģistrāciju (pārreģistrāciju), tām piešķir
reģistrācijas (pārreģistrācijas) numuru, kuru norāda
reģistrācijas apliecībā.
143.2 Zāļu
reģistrēšanas procesā vai pēc tam, kad ir pieņemts lēmums par
zāļu reģistrāciju, nosaka to piederību pie recepšu vai bezrecepšu
zālēm saskaņā ar Ministru kabineta noteikumiem par zāļu
klasifikāciju recepšu un bezrecepšu zālēs, kā arī ņemot vērā
Farmācijas likuma 28.1 pantā minētajos Eiropas
Komisijas ieteikumos (vadlīnijās) par dokumentāciju, ko Eiropas
Komisija ir publicējusi Eiropas Kopienas Zāļu tiesiskā regulējuma
2.sējumā, iekļautās zāļu klasificēšanas vadlīnijas.”
34. Izteikt 144. un 145.punktu
šādā redakcijā:
“144. Lēmumu par attiecīgo zāļu
reģistrācijas atteikumu vai atteikuma atcelšanu, reģistrācijas
apturēšanu vai apturēšanas atcelšanu, reģistrācijas anulēšanu vai
anulēšanas atcelšanu Valsts zāļu aģentūra ne vēlāk kā 10 dienu
laikā pēc lēmuma pieņemšanas izsniedz (nosūta ierakstītā vēstulē)
personai, uz kuru attiecas pieņemtais lēmums.
145. Lēmumu par attiecīgo zāļu
reģistrāciju vai pārreģistrāciju Valsts zāļu aģentūra paziņo
personai, uz kuru attiecas pieņemtais lēmums, un izsniedz
reģistrācijas apliecību, kas apliecina zāļu reģistrācijas vai
pārreģistrācijas faktu, ne vēlāk kā 30 dienu laikā pēc lēmuma
pieņemšanas. Valsts zāļu aģentūra noformē reģistrācijas apliecību
atbilstoši šo noteikumu 5.pielikumā noteiktajai veidlapai. Zāļu
reģistrācijas apliecība nav spēkā, ja ir pieņemts lēmums par
reģistrācijas anulēšanu vai reģistrācijas apturēšanu.”
35. Papildināt noteikumus ar
148.3.1 apakšpunktu šādā redakcijā:
“148.3.1 Valsts zāļu
aģentūra pēc zāļu reģistrācijas komisijas sēdes paziņo Zāļu cenu
aģentūrai par pieņemto lēmumu, lai Zāļu cenu aģentūra operatīvi
varētu izvērtēt informāciju par zālēm, kuras ir iekļautas vai
kuras paredzēts iekļaut kompensējamo zāļu sarakstā;”.
36. Papildināt noteikumus ar
148.5., 148.6. un 148.7.apakšpunktu šādā redakcijā:
“148.5. nodrošina, ka Eiropas Zāļu
novērtēšanas aģentūra tiek nekavējoties informēta par lēmumiem,
kas attiecas uz zāļu reģistrāciju saskaņā ar savstarpējās
atzīšanas procedūru vai reģistrācijas atteikumu, anulēšanu vai
anulēšanas atcelšanu, kā arī sniedz lēmumu pamatojumu. Valsts
zāļu aģentūra nodrošina atbilstošu informāciju par uzsāktajām
darbībām, kas var ietekmēt sabiedrības veselības aizsardzību
trešajās valstīs, un nosūta to Pasaules veselības organizācijai
un Eiropas Zāļu novērtēšanas aģentūrai;
148.6. dara zināmu citām
dalībvalstīm informāciju, kas nepieciešama Eiropas Kopienā ražoto
un izplatīto homeopātisko zāļu kvalitātes un drošības
garantēšanai, īpaši šo noteikumu 148.5.apakšpunktā minētajā
jautājumā;
148.7. publicē valsts valodā zāļu
reģistrācijas, pārreģistrācijas pieprasījuma un izmaiņu
apstiprināšanas veidlapu formas, ko Eiropas Komisija ir
publicējusi Eiropas Kopienas Zāļu tiesiskā regulējuma sējumos, un
konsultē reģistrācijas apliecības īpašniekus un pretendentus par
dokumentācijas noformēšanas prasībām, ko nosaka attiecīgie
Latvijas un Eiropas Kopienas tiesību akti, kā arī par Eiropas
Komisijas darba grupās izskatītiem jautājumiem.”
37. Svītrot 5., 16., 20., 28.,
139., 156., 160. un 163.punktu un XII nodaļu.
38. Aizstāt 166.punktā vārdus
“līdz īpašu Ministru kabineta noteikumu spēkā stāšanās dienai” ar
skaitļiem un vārdiem “līdz 2004 gada 30.aprīlim”.
39. Aizstāt 165.un 167.punktā
vārdus “īpašiem Ministru kabineta noteikumiem” ar skaitļiem un
vārdiem “2004.gada 1.maiju”.
40. Papildināt noteikumus ar 168.,
169. un 170.punktu šādā redakcijā:
“168. Valsts zāļu aģentūrai ir
tiesības atteikt zāļu reģistrāciju un pārreģistrāciju ar
2004.gada 1.maiju, ja zāļu reģistrācijas pieprasītājs nav
reģistrēts Eiropas Kopienā.
169. Šo noteikumu 5.pielikumā
noteiktais zāļu reģistrācijas apliecības noformējums neattiecas
uz zāļu reģistrācijas apliecībām, kas ir izsniegtas līdz šo
noteikumu spēkā stāšanās dienai.
170. Valsts zāļu aģentūra
atbilstoši Eiropas Komisijas ieteikumiem (vadlīnijām) par
dokumentāciju, ko Eiropas Komisija ir publicējusi Eiropas
Kopienas Zāļu tiesiskā regulējuma sējumos, ņem vērā, ka
dalībvalstīs jauniem reģistrācijas pieprasījumiem standartizētais
reģistrācijas dokumentācijas formāts stājās spēkā ar 2003.gada
31.decembri. Detalizētāka informācija par reģistrācijas
dokumentācijas formātu noteikta Eiropas Komisijas ieteikumos
(vadlīnijās) par dokumentāciju, ko Eiropas Komisija ir
publicējusi Eiropas Kopienas Zāļu tiesiskā regulējuma 2.sējumā,
kur noteikti gadījumi, kad atļauts iepriekšējais reģistrācijas
dokumentācijas noformējums (saskaņā ar šo noteikumu 26., 27.,
33., 34., 35., 37., 38., 39., 40., 41., 42. un 43.punktu un V,
VI, VII, VIII nodaļas prasībām).”
41. Aizstāt 49.4.3., 55.1.5.,
65.2., 135.2.apakšpunktā un 150., 151. un 167.punktā vārdu
“pielikums” (attiecīgā locījumā) ar skaitli un vārdu
“1.pielikums” (attiecīgā locījumā).
42. Izteikt informatīvu atsauci uz
Eiropas Savienības direktīvām šādā redakcijā:
“Informatīva
atsauce uz Eiropas Savienības direktīvām
Noteikumos iekļautas tiesību
normas, kas izriet no:
1) Eiropas Parlamenta un Padomes
2001.gada 6.novembra Direktīvas 2001/83/EK par Kopienas kodeksu,
kas attiecas uz cilvēkiem paredzētām zālēm;
2) Komisijas 2003.gada 25.jūlija
Direktīvas 2003/63/EK (2003.gada 25.jūnijs), ar ko groza Eiropas
Parlamenta un Padomes Direktīvu 2001/83/EK par Kopienas kodeksu,
kas attiecas uz cilvēkiem paredzētām zālēm.”
43. Papildināt noteikumus ar 2.,
3., 4. un 5.pielikumu šādā redakcijā:
“2.pielikums
Ministru kabineta
2000.gada 31.oktobra
noteikumiem Nr.381
Analīzes,
farmakotoksikoloģijas un klīniskie standarti un protokoli
attiecībā uz zāļu testēšanu
A sadaļa
Standartizētās reģistrācijas dokumentācijas prasības
I nodaļa
1.modulis. Administratīvā informācija
1. Sniedz iesniegtā reģistrācijas
pieprasījuma dokumentācijas 1., 2., 3., 4. un 5.moduļa satura
rādītāju.
2. Pieprasījuma veidlapa:
(1) Zāles, uz kurām attiecas
pieprasījums, identificē, norādot nosaukumu un aktīvās(-o)
vielas(-u) nosaukumu, kā arī zāļu formu, lietošanas veidu,
stiprumu un galīgo noformējumu (arī iepakojumu).
(2) Norāda pieprasījuma
iesniedzēja nosaukumu un adresi, ražotāju nosaukumus un adreses,
kā arī objektus, kas iesaistīti dažādos ražošanas posmos, arī
galaprodukta ražotāju un aktīvās(-o) vielas(-u) ražotāju(-us), ja
nepieciešams, — importētāja nosaukumu un adresi.
(3) Pieprasījuma iesniedzējs
norāda pieprasījuma veidu, kā arī iesniegtos paraugus, ja tādi
ir.
(4) Administratīvo datu pielikumā
iekļauj zāļu ražošanas atļaujas (licences) kopijas un to valstu
sarakstu, kuras piešķīrušas minēto atļauju, dalībvalstu
apstiprināto zāļu aprakstu kopijas saskaņā ar šo noteikumu
36.punktu, un to valstu sarakstu, kurām iesniegts reģistrācijas
pieprasījums.
(5) Pieprasījuma iesniedzēji,
inter alia, sniedz detalizētu informāciju par zālēm, uz
kurām attiecas reģistrācijas pieprasījums, pieprasījuma juridisko
pamatu, paredzamo reģistrācijas turētāju (turpmāk —
reģistrācijas īpašnieks) un ražotāju(-us), kā arī informāciju par
to, kāds ir zāļu statuss reti sastopamu slimību ārstēšanai,
zinātnisku informāciju un pediatrijas attīstības programmu.
3. Zāļu apraksts, marķējums un
lietošanas instrukcija:
(1) Reģistrācijas pieprasījuma
iesniedzējs piedāvā zāļu aprakstu saskaņā ar šo noteikumu
36.punktu.
(2) Reģistrācijas pieprasījuma
iesniedzējs norāda marķējuma tekstu primārajam un sekundārajam
iepakojumam, kā arī lietošanas instrukcijai. Marķējuma teksts
atbilst šādām prasībām:
1) informācija marķējumā un zāļu
lietošanas instrukcijā, kā arī tajā lietotie simboli vai
piktogrammas atbilst normatīvo aktu prasībām par zāļu marķēšanas
kārtību un zāļu lietošanas instrukcijas prasībām. Informācija ir
tās dalībvalsts oficiālajā valodā vai valodās, kurā zāles
izplata;
2) lietošanas instrukcija ir
uzrakstīta tā, lai lietotājiem tā būtu skaidra un saprotama;
3) marķējumā un lietošanas
instrukcijā ir ievērotas Farmācijas likuma 28.1 pantā
minētajos Eiropas Komisijas ieteikumos (vadlīnijās) par
dokumentāciju, ko Eiropas Komisija publicējusi Eiropas Kopienas
Zāļu tiesiskā regulējuma 2.sējumā, noteiktās prasības marķējuma
un lietošanas instrukcijas salasāmībai;
4) atļauts marķējumā norādīt
datus, kā arī iespiest lietošanas instrukciju vairākās valodās,
ja visās valodās sniedz vienus un tos pašus datus un
informāciju.
(3) Reģistrācijas pieprasījuma
iesniedzējs sniedz attiecīgo zāļu primārā un sekundāra
iepakojuma, marķējuma (etiķešu) un lietošanas instrukciju
paraugus vai maketus.
(4) Pieprasījuma veidlapas
administratīvo datu pielikumā, ja nepieciešams, iekļauj
dalībvalstu jau apstiprināto zāļu aprakstu kopijas saskaņā ar šo
noteikumu 36., 137. un 138.punktu un to valstu sarakstu, kurām
iesniegts reģistrācijas pieprasījums.
4. Informācija par ekspertiem:
(1) Saskaņā ar šo noteikumu 13. un
14.punktu eksperti iesniedz detalizēti izstrādātus novērojumu
ziņojumus par dokumentiem un datiem, kuri veido reģistrācijas
dokumentāciju, jo īpaši par 3., 4. un 5.moduli (attiecīgi
ķīmiskā, farmaceitiskā un bioloģiskā dokumentācija, neklīniskās
izpētes dokumentācija un klīniskās izpētes dokumentācija).
Eksperti pievērš uzmanību kritiskajiem punktiem, kas saistīti ar
zāļu kvalitāti, un pētījumiem, kuri veikti ar dzīvniekiem un
cilvēkiem, kā arī norāda visus novērtējumam svarīgos datus.
(2) Minētās prasības izpilda,
sniedzot vispārēju kvalitātes pārskatu, neklīniskās izpētes
pārskatu (dati no pētījumiem, kas veikti ar dzīvniekiem) un
klīniskās izpētes pārskatu, ko pievieno reģistrācijas
dokumentācijas 2.modulim. Ekspertu parakstītu deklarāciju kopā ar
īsu informāciju par viņu izglītību, apmācību un profesionālo
pieredzi sniedz 1.modulī. Ekspertiem nepieciešama piemērota
tehniskā vai profesionālā kvalifikācija. Jāatspoguļo eksperta un
reģistrācijas pieprasījuma iesniedzēja profesionālās
attiecības.
5. Īpašas prasības atšķirīgiem
reģistrācijas pieprasījumiem ir sniegtas II nodaļā.
6. Vides riska novērtējums:
(1) Ja nepieciešams, reģistrācijas
pieprasījumos iekļauj riska novērtējuma pārskatu, kurā novērtēts
iespējamais zāļu lietošanas, kā arī iznīcināšanas risks videi, un
sniedz priekšlikumus attiecīga marķējuma noteikumiem. Jāpievērš
uzmanība vides riskam, kas saistīts ar tādu zāļu izplatīšanu
vidē, kas satur ģenētiski modificētus organismus atbilstoši
normatīviem par ģenētiski modificētu organismu apzinātu
izplatīšanu vidē.
(2) Informāciju, kas attiecas uz
vides risku, pievieno 1.moduļa papildinājumā.
(3) Informāciju sniedz saskaņā ar
minētajiem normatīviem, ņemot vērā Eiropas Komisijas publicētās
vadlīnijas par to īstenošanu.
(4) Informācijā ietilpst:
1) ievads;
2) rakstiska piekrišana (vai
piekrišanas kopija) par ģenētiski modificētu organismu apzinātu
izplatīšanu vidē izpētes un attīstības nolūkā saskaņā ar
normatīviem;
3) informācija par ģenētiski
modificētu organismu atklāšanas un identificēšanas metodēm un
kodu, kā arī vides riska novērtēšanai svarīga papildinformācija
par ģenētiski modificētiem organismiem vai produktu saskaņā ar
normatīviem;
4) vides riska novērtējums, kas
sagatavots, pamatojoties uz normatīvos noteikto informāciju;
5) ņemot vērā iepriekšminēto
informāciju un vides riska novērtējumu, atzinums, kurā ieteikta
atbilstoša riska pārvaldības stratēģija un ietverts plāns
attiecīgo pārdoto ģenētiski modificēto organismu un produktu
uzraudzībai pēc laišanas tirgū, un norādīti īpašie dati, kuriem
jābūt iekļautiem zāļu aprakstā, marķējumā un lietošanas
instrukcijā;
6) attiecīgie sabiedrības
informēšanas pasākumi.
(5) Nepieciešams autora paraksts
un datums, kā arī informācija par autora izglītību, apmācību un
profesionālo pieredzi un paziņojums par autora attiecībām ar
reģistrācijas pieprasījuma iesniedzēju.
II nodaļa
2.modulis. Kopsavilkumi
7. Šī moduļa mērķis ir apkopot
ķīmiskos, farmaceitiskos un bioloģiskos datus, neklīniskās
izpētes datus un klīniskās izpētes datus, kas noteikti
reģistrācijas dokumentācijas 3., 4. un 5.modulī, kā arī sniegt šo
noteikumu 13. un 14.punktā minētos ziņojumus un pārskatus.
8. Uzmanību pievērš kritiskajiem
punktiem un analizē tos. Sniedz faktu kopsavilkumus (arī tabulā).
Ziņojumos sniedz savstarpējas norādes uz tabulām vai uz
informāciju, kas ietverta 3.moduļa (ķīmiskā, farmaceitiskā un
bioloģiskā dokumentācija), 4.moduļa (neklīniskās izpētes
dokumentācija) un 5.moduļa (klīniskās izpētes dokumentācija)
galvenajā dokumentācijā. Informāciju, kas ietverta 2.modulī,
sniedz, ievērojot formu, saturu un numerācijas sistēmu, kas
norādīta 2.sējumā par paziņojumu reģistrācijas pieprasījuma
iesniedzējiem. Pārskati un kopsavilkumi atbilst tālākminētajiem
pamatprincipiem un pamatprasībām.
9. Kopējais satura rādītājs
2.modulī iekļauj 2., 3., 4. un 5.modulī iesniegtās zinātniskās
dokumentācijas satura rādītāju.
10. Ievadā sniedz informāciju par
to zāļu farmakoloģisko klasi, darbības veidu un paredzēto
klīnisko izmantošanu, kurām pieprasa reģistrāciju.
11. Vispārējā kvalitātes
kopsavilkumā sniedz pārskatu par ķīmiskajiem, farmaceitiskajiem
un bioloģiskajiem datiem. Uzsver galvenos kritiskos parametrus un
ar kvalitāti saistītos jautājumus, kā arī pamatojumu, ja netiek
ievērotas attiecīgās pamatnostādnes. Šajā dokumentā ievērota
3.modulī sniegto detalizēto datu joma un izklāsts.
12. Neklīniskās izpētes
pārskats:
(1) Nepieciešams integrēti un
kritiski novērtēt zāļu neklīnisko izpēti ar dzīvniekiem (in
vitro). Izklāsta un pamato testēšanas stratēģiju un atkāpes
no attiecīgajām pamatnostādnēm.
(2) Neklīniskās izpētes pārskatā
iekļauj piemaisījumu un sadalīšanās produktu un to iespējamās
farmakoloģiskās un toksikoloģiskās iedarbības novērtējumu
(izņemot bioloģiskas izcelsmes zālēm). Izskata hilaritātes,
ķīmiskās formas un piemaisījumu sastāva atšķirības starp
neklīniskajā izpētē izmantoto savienojumu un tirgū izplatāmajām
zālēm.
(3) Novērtē bioloģiskas izcelsmes
zāļu neklīniskajā izpētē un klīniskajā izpētē izmantotā materiāla
un pārdodamo zāļu salīdzināmību.
(4) Jaunās palīgvielas īpaši
novērtē drošības ziņā.
(5) Nosaka zāļu īpašības
atbilstoši neklīniskajai izpētei, kā arī izskata iegūtos datus
par zāļu drošību paredzētajā klīniskajā izmantojumā
cilvēkiem.
13. Klīniskās izpētes
pārskats:
(1) Klīniskās izpētes pārskats
paredzēts, lai sniegtu klīniskajā kopsavilkumā un 5.modulī
iekļauto klīnisko datu kritisku analīzi. Paredz zāļu klīniskās
izstrādes metodi, arī kritisku pētījuma plānu, un ar pētījumiem
saistītos lēmumus un pētījumu veikšanu.
(2) Klīniskās izpētes pārskatā
sniedz īsu pārskatu par iegūtajiem klīniskajiem datiem, tajā
skaitā par būtiskiem ierobežojumiem, kā arī ieguvumu un risku
novērtējumu, pamatojoties uz klīnisko pētījumu secinājumiem.
Vajadzīgs skaidrojums par to, kā iegūtie dati par efektivitāti un
drošību pamato paredzēto devu un mērķindikācijas, kā arī
novērtējums par to, kā zāļu apraksts un citi paņēmieni optimizēs
ieguvumus un pārvaldīs riskus.
(3) Izskaidro efektivitātes un
drošības jautājumus, kas radušies zāļu izstrādes laikā, kā arī
neatrisinātos jautājumus.
14. Neklīniskās izpētes
kopsavilkumā ar dzīvniekiem (in vitro) veikto
farmakoloģijas, farmakokinētikas un toksikoloģijas pētījumu
rezultātus sniedz rakstiski kā faktu (arī tabulā), ko iesniedz
šādā secībā:
(1) Ievads.
(2) Rakstisks kopsavilkums par
farmakoloģiju.
(3) Kopsavilkums par farmakoloģiju
tabulā.
(4) Rakstisks kopsavilkums par
farmakokinētiku.
(5) Kopsavilkums par
farmakokinētiku tabulā.
(6) Rakstisks kopsavilkums par
toksikoloģiju.
(7) Kopsavilkums par toksikoloģiju
tabulā.
15. Klīniskās izpētes kopsavilkumā
sniedz detalizētu faktu kopsavilkumu par zāļu klīnisko
informāciju, kas iekļauta 5.modulī. Tajā iekļauj biofarmaceitisko
pētījumu, klīniskās farmakoloģijas pētījumu un klīniskās
efektivitātes un drošības pētījumu rezultātus. Nepieciešams
atsevišķo pētījumu konspekts. Apkopoto informāciju par klīnisko
izpēti iesniedz šādā secībā:
(1) Kopsavilkums par
biofarmaceitiskajiiem pētījumiem un saistītajām analīzes
metodēm.
(2) Klīnisko farmakoloģijas
pētījumu kopsavilkums.
(3) Kopsavilkums par klīnisko
efektivitāti.
(4) Kopsavilkums par klīnisko
drošību.
(5) Atsevišķo pētījumu
konspekts.
III nodaļa
3.modulis. Ķīmiskā, farmaceitiskā un bioloģiskā informācija par
zālēm, kas satur gan ķīmiskas, gan bioloģiski aktīvas vielas
16. Forma un noformējums.
Vispārīgs 3.moduļa izklāsts ir šāds:
16.1. satura rādītājs;
16.2. datu kopums:
16.2.1. aktīvā viela:
(1) Vispārīga informācija:
1) nomenklatūra;
2) struktūra;
3) vispārīgas īpašības.
(2) Ražošana:
1) ražotājs(-i);
2) ražošanas procesa un procesa
vadības apraksts;
3) izejvielu kontrole;
4) kritisko posmu un starpposmu
pārbaudes;
5) procesa validācija un/vai
novērtējums;
6) ražošanas procesa
uzlabošana.
(3) Raksturojums:
1) struktūras un citu raksturīgo
pazīmju izskaidrojums;
2) piemaisījumi.
(4) Aktīvās vielas kontrole:
1) specifikācija;
2) analītiskās procedūras;
3) analītisko procedūru
validācija;
4) sēriju analīzes;
5) specifikācijas pamatojums.
(5) References standarti vai
materiāli.
(6) Iepakojuma noslēgšanas
sistēma.
(7) Stabilitāte:
1) kopsavilkums un secinājumi par
stabilitāti;
2) stabilitātes protokols (pēc
apstiprināšanas) un ar stabilitāti saistītās saistības;
3) dati par stabilitāti;
16.2.2. gatavās zāles:
(1) Zāļu apraksts un sastāvs.
(2) Farmaceitiskā izstrāde:
1) zāļu sastāvdaļas:
a) aktīvā viela;
b) palīgvielas;
2) zāles:
a) preparāta izstrāde;
b) devas palielināšana;
c) fizikālķīmiskās īpašības;
d) bioloģiskās īpašības;
3) ražošanas procesa
uzlabošana;
4) iepakojuma noslēgšanas
sistēma;
5) mikrobioloģiskās īpašības;
6) saderība.
(3) Ražošana:
1) ražotājs(-i);
2) sērijas sastāvs;
3) ražošanas procesa un procesa
vadības apraksts;
4) kritisko posmu un starpposmu
pārbaudes;
5) procesa validācija un/vai
novērtējums.
(4) Palīgvielu kontrole:
1) specifikācijas;
2) analītiskās procedūras;
3) analītisko procedūru
validācija;
4) specifikāciju pamatojums;
5) cilvēku vai dzīvnieku izcelsmes
palīgvielas;
6) jaunas palīgvielas.
(5) Gatavo zāļu kontrole:
1) specifikācija(-as);
2) analītiskās procedūras;
3) analītisko procedūru
validācija;
4) sēriju analīzes;
5) piemaisījumu raksturojums;
6) specifikācijas(-u)
pamatojums.
(6) References standarti vai
materiāli.
(7) Iepakojuma noslēgšanas
sistēma.
(8) Stabilitāte:
1) kopsavilkums un secinājumi par
stabilitāti;
2) stabilitātes protokols (pēc
apstiprināšanas) un ar stabilitāti saistītās saistības;
3) dati par stabilitāti;
16.2.3. papildinājumi:
(1) Aprīkojums un iekārtas (tikai
bioloģiskas izcelsmes zālēm).
(2) Iespējamā slimības
ierosinātāja drošības novērtējums.
(3) Palīgvielas;
16.2.4. Eiropas Kopienas papildu
informācija:
(1) Zāļu ražošanas procesa
validācijas shēma.
(2) Medicīnas ierīce.
(3) Atbilstības
sertifikāts(-i).
(4) Zāles, kuras satur vai kuru
ražošanas procesā izmanto dzīvnieku un/vai cilvēka izcelsmes
izejvielas (TSE procedūra);
16.3. literatūras atsauces.
17. Saturam ir šādi pamatprincipi
un pamatprasības:
17.1. ķīmiskajos, farmaceitiskajos
un bioloģiskajos datos iekļauj šādu informāciju par aktīvo(-ajām)
vielu(-ām) un par gatavajām zālēm:
17.1.1. izstrāde;
17.1.2. ražošanas process;
17.1.3. raksturojums un
īpašības;
17.1.4. kvalitātes kontroles
darbības un prasības;
17.1.5. stabilitāte;
17.1.6. gatavo zāļu sastāva un
noformējuma apraksts;
17.2. sniedz divas galvenās
informācijas paketes attiecīgi par
aktīvo(-ajām) vielu(-ām) un par
gatavajām zālēm;
17.3. šajā modulī papildus sniedz
detalizētu informāciju par izejvielām un neapstrādātiem
materiāliem, kurus izmanto aktīvās(-o) vielas(-u) ražošanā, un
par palīgvielām, kuras iekļauj gatavajās zālēs;
17.4. procedūras un metodes, kuras
izmanto aktīvās vielas un gatavo zāļu ražošanā un kontrolē,
detalizēti apraksta, lai tās varētu atkārtot pārbaudēs, kuras
veic pēc kompetentās iestādes (Latvijā — Valsts zāļu
aģentūras) pieprasījuma. Analīzes procedūras atbilst jaunākajām
zinātnes atziņām un ir validētas. Tiek sniegti validācijas
pētījumu rezultāti. Veicot Eiropas Farmakopejā iekļautās testu
procedūras, šo aprakstu aizstāj ar atbilstošo detalizēto atsauci
uz monogrāfiju(-ām) un vispārējo(-ējām) nodaļu(-ām);
17.5. Eiropas Farmakopejas
monogrāfijas attiecas uz tajā iekļautajām vielām, preparātiem un
zāļu formām. Attiecībā uz citām vielām katra dalībvalsts var
prasīt, lai tiktu ievērota tās nacionālā farmakopeja.
(1) Ja Eiropas Farmakopejā vai
dalībvalsts farmakopejā minētais materiāls ir pagatavots ar tādu
metodi, kuras dēļ tajā var palikt piemaisījumi, uz kuriem
neattiecas farmakopejas monogrāfija, jānorāda šie piemaisījumi un
to maksimālais pieļaujamais daudzums un jāapraksta piemērota
analīzes procedūra.
(2) Ja Eiropas Farmakopejā vai
dalībvalsts farmakopejā iekļautā specifikācija varētu nebūt
pietiekama, lai nodrošinātu vielas kvalitāti, kompetentās
iestādes (Latvijā — Valsts zāļu aģentūra) var pieprasīt no
reģistrācijas īpašnieka piemērotākas specifikācijas. Kompetentās
iestādes (Latvijā — Valsts zāļu aģentūra) informē par
attiecīgo farmakopeju atbildīgās iestādes. Reģistrācijas
īpašnieks sniedz šo farmakopeju pārzinošajām iestādēm detalizētas
ziņas par varbūtējo nepilnību un par papildus izmantotajām
specifikācijām.
(3) Veicot Eiropas Farmakopejā
iekļautās analītiskās procedūras, šo aprakstu katrā attiecīgajā
sadaļā aizstāj ar atbilstošo detalizēto atsauci uz
monogrāfiju(-ām) un vispārējo(-ām) nodaļu(-ām);
17.6. ja izejvielas un
neapstrādāti materiāli, aktīvā(-ās) viela(-as) vai
palīgviela(-as) nav aprakstīti ne Eiropas Farmakopejā, ne
dalībvalsts farmakopejā, var akceptēt atbilstību monogrāfijai
kādas trešās valsts (valsts, kura nav Eiropas Savienības
dalībvalsts vai Eiropas brīvās tirdzniecības asociācijas valsts
(EBTA valsts), kas parakstījusi Eiropas Ekonomikas zonas līgumu)
farmakopejā. Šādos gadījumos reģistrācijas pieprasījuma
iesniedzējs iesniedz monogrāfijas eksemplāru kopā ar monogrāfijā
ietverto analītisko procedūru validējumu un, ja
nepieciešams, — tulkojumu;
17.7. ja aktīvā viela,
neapstrādātais materiāls, kā arī izejviela(-as) vai
palīgviela(-as) ir iekļauta(-as) kādā Eiropas Farmakopejas
monogrāfijā, reģistrācijas pieprasījuma iesniedzējs var pieprasīt
atbilstības sertifikātu, kuru norāda attiecīgajā šī moduļa
sadaļā, ja to piešķīris Eiropas Zāļu kvalitātes direktorāts.
Uzskata, ka Eiropas Farmakopejas monogrāfijas atbilstības
sertifikāti aizstāj attiecīgos šajā modulī aprakstīto iedaļu
datus. Ražotājs reģistrācijas pieprasījuma iesniedzējam sniedz
rakstisku apstiprinājumu, ka ražošanas process nav mainīts, kopš
Eiropas Zāļu kvalitātes direktorāts izdevis atbilstības
sertifikātu;
17.8. aktīvās vielas ražotājs vai
reģistrācijas pieprasījuma iesniedzējs var vienoties, ka
informāciju par noteiktu aktīvo vielu ražošanas procesa
detalizētu aprakstu, kvalitātes kontroli ražošanas laikā un
procesa validāciju aktīvās vielas ražotājs iesniedz atsevišķā
dokumentā kā aktīvās vielas pamatlietu tieši kompetentajām
iestādēm (Latvijā — Valsts zāļu aģentūra). Šajā gadījumā ražotājs
sniedz reģistrācijas pieprasītājam ziņas, kas iesniedzējam var
būt vajadzīgas, lai varētu uzņemties atbildību par attiecīgajām
zālēm. Ražotājs rakstiski apstiprina reģistrācijas pieprasījuma
iesniedzējam, ka viņš nodrošina sēriju atbilstību paraugam un bez
reģistrācijas pieprasītāja ziņas nemaina ražošanas procesu vai
specifikācijas. Kompetentajās iestādēs (Latvijā — Valsts zāļu
aģentūra) iesniedz dokumentus un ziņas, kas pamato reģistrācijas
pieprasījumu par minētajām izmaiņām. Minētos dokumentus un ziņas
iesniedz arī reģistrācijas pieprasījuma iesniedzējam, izskatot
aktīvās vielas pamatlietas atklāto daļu;
17.9. veicot īpašus pasākumus, kas
saistīti ar dzīvnieku sūkļveida encefalopātijas pārnešanas
novēršanu, reģistrācijas pieprasījuma iesniedzējam jāpierāda
katrā ražošanas procesa posmā izmantoto materiālu atbilstība
“Norādījumiem dzīvnieku sūkļveida encefalopātijas ierosinātāju
izplatīšanas riska samazināšanai ar zālēm” un to atjauninātajām
redakcijām, ko Eiropas Komisija publicējusi Eiropas Savienības
Oficiālajā Vēstnesī. Atbilstību minētajiem norādījumiem var
pierādīt, iesniedzot Eiropas Zāļu kvalitātes direktorātā
sertifikātu par atbilstību attiecīgajai Eiropas Farmakopejas
monogrāfijai vai arī sniedzot zinātniskus datus, kas šo
atbilstību pamato;
17.10. par iespējamajiem slimības
ierosinātājiem sniedz informāciju, kurā novērtēts iespējamo
vīrusu vai nevīrusu ierosinātāju piemaisījumu risks, kā noteikts
attiecīgajās pamatnostādnēs, kā arī attiecīgajā Eiropas
Farmakopejas vispārīgajā monogrāfijā un vispārīgajā nodaļā;
17.11. pietiekami detalizēti
apraksta zāļu ražošanas procesa posmus un pārbaudēs izmantojamās
speciālās iekārtas un aprīkojumu;
17.12. ja nepieciešams, sniedz CE
marķējumu, kas prasīts Eiropas Kopienas tiesību aktos par
medicīnas ierīcēm;
17.13. īpašu uzmanību pievērš
šādiem atsevišķiem elementiem:
17.13.1. aktīvā(-ās)
viela(-as):
(1) Vispārīga informācija un
informācija par izejvielām un neapstrādātiem materiāliem:
1) sniedz informāciju par aktīvās
vielas nomenklatūru, arī ieteikto starptautisko nepatentēto
nosaukumu, Eiropas Farmakopejas nosaukumu, ja nepieciešams,
ķīmisko(-os) nosaukumu(-us). Norāda struktūrformulu, arī relatīvo
un absolūto stereoķīmiju, molekulformulu un relatīvo molekulmasu.
Biotehnoloģiskām zālēm piemērotos gadījumos norāda shematisko
aminoskābju sekvenci un relatīvo molekulmasu. Sniedz sarakstu ar
aktīvās vielas fizikālķīmiskajām un citām īpašībām. Bioloģiskas
izcelsmes zālēm norāda bioloģisko aktivitāti;
2) šī pielikuma izpratnē
izejvielas ir materiāli, no kā ražo vai ekstrahē aktīvo vielu.
Attiecībā uz bioloģiskas izcelsmes zālēm izejvielas ir
bioloģiskas izcelsmes vielas (piemēram, mikroorganismi, augu vai
dzīvnieku orgāni un audi, cilvēku vai dzīvnieku izcelsmes šūnas
vai šķidrumi (arī asinis vai plazma)) un šūnu biotehnoloģiskā
uzbūve (šūnu substrāti, kas ir rekombinanti vai nav tādi, arī
cilmes šūnas). Bioloģiskās izcelsmes zāles ir preparāts, kurā
aktīvā viela ir bioloģiska viela. Bioloģiskas izcelsmes viela ir
tāda viela, kuru ražo vai ekstrahē no bioloģiska avota un kuras
raksturojumam un kvalitātes noteikšanai jāveic fizikālas,
ķīmiskas un bioloģiskas pārbaudes, kā arī jākontrolē ražošanas
process. Citas vielas, ko izmanto aktīvās(-o) vielas(-u) ražošanā
vai ekstrahēšanā, bet no kurām tieši šo aktīvo vielu neiegūst
(piemēram, reaģenti, barotnes, teļu embrionālais serums,
piedevas, hromatogrāfijā izmantojamās bufervielas), sauc par
neapstrādātiem materiāliem. Par bioloģiskas izcelsmes zālēm
uzskata:
a) imunoloģiskus preparātus (
jebkuras zāles, ko veido vakcīnas, toksīni, serumi vai alergēni)
;
b) no cilvēka asins un plazmas
iegūtas zāles ( valsts vai privātu uzņēmumu rūpnieciski
izgatavotas zāles, kuru pamatā ir asins komponenti, tajā skaitā
tādas zāles kā cilvēku albumīns, asinsreces faktori un
imunoglobulīni);
c) zāles, kuras nevar laist
Eiropas Kopienas tirgū, ja Eiropas Kopiena nav izsniegusi
tirdzniecības atļauju saskaņā ar Padomes 1993.gada 22.jūnija
Regulu (EEK) Nr.2309/03, ar ko nosaka Eiropas Kopienas procedūru
tādu zāļu apstiprināšanai un pārraudzībai, kuras paredzētas
izmantošanai cilvēkiem un veterinārijā, un ar ko nodibina Eiropas
Zāļu novērtēšanas aģentūru;
d) šajā pielikumā definētās
uzlabotās terapeitiskās iedarbības zāles;
3) vakcīnas, toksīni vai serumi
īpaši attiecas uz:
a) ierosinātājiem, ko izmanto
aktīvās imunitātes radīšanā (piemēram, holēras vakcīnu,
Kalmeta-Gerēna prettuberkulozes vakcīnu bcg, poliomielīta
vakcīnām, baku vakcīnu) ;
b) ierosinātājiem, ko izmanto
imunitātes stāvokļa noteikšanā, tajā skaitā tuberkulīnam un PPD
tuberkulīniem, toksīniem Šika testam un Dika testam,
brucelīnam;
c) ierosinātājiem, ko izmanto
pasīvās imunitātes radīšanā (piemēram, difterijas antitoksīnu,
pretbaku globulīnu, pretlimfocītu globulīnu);
4) alergēni ir zāles, kas
paredzētas specifisku iegūtu pārmaiņu noteikšanai vai
izraisīšanai imunoloģiskajā atbildē uz alerģijas
ierosinātāju.
(2) Aktīvās(-o) vielas(-u)
ražošanas process:
1) tā apraksts nosaka
reģistrācijas pieprasījuma iesniedzēja saistības ražot aktīvo
vielu. Lai pienācīgi aprakstītu ražošanas procesu un procesa
vadību, sniedzama atbilstoša informācija, kas noteikta Eiropas
Zāļu novērtēšanas aģentūras publicētajās pamatnostādnēs;
2) uzskaita aktīvās(-o) vielas(-u)
ražošanai nepieciešamos materiālus, norādot, kā katru materiālu
izmanto procesā. Sniedz informāciju par šo materiālu kvalitāti un
pārbaudi. Sniedz informāciju, kas pierāda, ka materiāli atbilst
paredzētās izmantošanas standartiem. Uzskaita neapstrādātos
materiālus, kā arī dokumentē to kvalitāti un pārbaudes. Norāda
katra ražotāja (arī līgumslēdzēju) un katras paredzētās ražošanas
vietas vai ražošanā un testēšanā iesaistītā objekta nosaukumu,
adresi un atbildību;
3) uz bioloģiskas izcelsmes zālēm
attiecas šādas papildu prasības:
a) aprakstīta un dokumentēta
izejmateriālu izcelsme un vēsture;
b) attiecībā uz īpašiem pasākumiem
dzīvnieku sūkļveida encefalopātijas pārnešanas novēršanai
reģistrācijas pieprasījuma iesniedzējam jāpierāda, ka aktīvā
viela atbilst “Norādījumiem dzīvnieku sūkļveida encefalopātijas
ierosinātāju izplatīšanas riska samazināšanai ar zālēm” un to
atjauninātajām redakcijām, ko Eiropas Komisija publicējusi
Eiropas Savienības Oficiālajā Vēstnesī;
c) ja izmanto šūnu bankas, norāda,
ka šūnu īpašības palikušas nemainītas pirms un pēc ražošanā
lietotās pasāžas;
d) pārbauda, vai sējmateriālos,
šūnu bankās, serumu vai plazmas fondos un citos bioloģiskas
izcelsmes materiālos un, ja iespējams, materiālos, no kuriem tie
iegūti, nav iespējamo slimības ierosinātāju;
e) ja potenciāli patogēnu
iespējamo slimības ierosinātāju klātbūtne ir nenovēršama,
attiecīgo materiālu izmanto tikai tad, ja papildu apstrāde
nodrošina to iznīcināšanu un inaktivāciju, un tam ir vajadzīga
validācija;
f) vakcīnas ražošanu pēc iespējas
balsta uz sējmateriālu sēriju sistēmu un uz izveidotajām šūnu
bankām. Bakteriālajām un vīrusu vakcīnām infekcijas izraisītāja
raksturīgās pazīmes pierāda sējmateriālā. Dzīvām vakcīnām
sējmateriālā papildus pierāda vājināšanās raksturīgo pazīmju
stabilitāti. Ja ar šo pierādījumu nepietiek, vājināšanās
raksturīgās pazīmes pierāda arī ražošanas posmā;
g) zālēm, kas iegūtas no cilvēka
asins vai plazmas, saskaņā ar šī pielikuma IV nodaļā noteiktiem
noteikumiem apraksta un dokumentē izejvielas izcelsmi, kritērijus
un savākšanas, transportēšanas un uzglabāšanas procedūras;
h) apraksta ražošanas iekārtas un
aprīkojumu;
4) ja nepieciešams, sniedz
informāciju par katrā kritiskajā posmā veiktajiem testiem un
pieņemšanas kritērijiem, starpsavienojumu kvalitāti un kontroli,
kā arī procesa validāciju un novērtējumu;
5) ja potenciāli patogēnu
iespējamo slimības ierosinātāju klātbūtne ir nenovēršama,
attiecīgo materiālu izmanto tikai tad, ja turpmākā pārstrāde
nodrošina tā iznīcināšanu un inaktivāciju, un tam ir vajadzīga
validācija iedaļā par vīrusu drošības novērtējumu;
6) apraksta un izskaidro tās
nozīmīgās izmaiņas, kas aktīvās vielas izstrādes laikā izdarītas
attiecībā uz ražošanas procesu un ražošanas vietu.
(3) Aktīvās(-o) vielas(-u)
raksturojums:
1) sniedz datus par aktīvās(-o)
vielas(-u) struktūru un citām raksturīgajām pazīmēm;
2) sniedz apstiprinājumu par
aktīvās(-o) vielas(-u) struktūru, pamatojoties uz
fizikālķīmiskajām vai imūnķīmiskajām, vai bioloģiskajām metodēm,
kā arī sniedz informāciju par piemaisījumiem.
(4) Aktīvās(-o) vielas(-u)
kontrole:
1) sniedz detalizētu informāciju
par specifikācijām, ko izmanto
aktīvās(-o) vielas(-u) regulārai
kontrolei, pamatojot šo specifikāciju izvēli, kā arī sniedz
informāciju par analīzes metodēm un to validāciju;
2) atspoguļo atsevišķu izstrādes
laikā ražotu sēriju pārbaužu rezultātus.
(5) Nosaka un detalizēti apraksta
references preparātus un references standartus. Ja nepieciešams,
izmanto Eiropas Farmakopejas ķīmisko un bioloģisko references
materiālu.
(6) Sniedz aktīvās vielas
iepakojuma un noslēgšanas sistēmas(-u) aprakstu un to
specifikācijas.
(7) Aktīvās(-o) vielas(-u)
stabilitāte:
1) apkopo veiktos pētījumus,
izmantotos protokolus un pētījumu rezultātus;
2) atbilstošā formā detalizēti
atspoguļo stabilitātes pētījumu rezultātus, iekļaujot informāciju
par datu sagatavošanai izmantotajām analītiskajām procedūrām un
par šo procedūru validāciju;
3) iesniedz stabilitātes protokolu
(pēc apstiprināšanas) un ar stabilitāti saistītās saistības;
17.13.2. gatavās zāles:
(1) Gatavo zāļu apraksts un
sastāvs:
1) iesniedz gatavo zāļu aprakstu
un norāda to sastāvu. Informācijā ietver zāļu formas un sastāva
aprakstu, norādot gatavo zāļu komponentus, to daudzumu vienībā un
šo komponentu funkcijas:
a) aktīvajai(-ajām)
vielai(-ām);
b) palīgvielu komponentiem
neatkarīgi no to īpašībām vai lietotā daudzuma (piemēram,
krāsvielām, konservantiem, palīgvielām, stabilizatoriem,
biezinātājiem, emulgatoriem, aromatizētājiem un aromātiskajām
vielām);
c) zāļu ārējā apvalka
komponentiem, kas paredzēti pacientam norīšanai vai citādai
lietošanai (piemēram, cietās kapsulas, mīkstās kapsulas,
supozitoriji, apvalkotas tabletes, tabletes plēves iepakojumā).
Minētajām ziņām pievieno attiecīgos datus par iepakojuma veidu
un, ja nepieciešams, par tā aizvēršanas veidu, kā arī datus par
zāļu lietošanas un ievadīšanas ierīcēm, ko piegādā kopā ar
zālēm;
2) šo noteikumu 18.3.apakšpunktā
noteikto zāļu kvalitatīvo un kvantitatīvo sastāvu norāda
vienkāršā terminoloģijā, kas zāļu komponentu aprakstīšanai
ir:
a) galvenais nosaukums attiecīgās
monogrāfijas augšdaļā, ar atsauci uz attiecīgo farmakopeju —
attiecībā uz vielām, kas ierakstītas Eiropas Farmakopejā vai, ja
tā nav, kādas dalībvalsts farmakopejā;
b) Pasaules veselības
organizācijas ieteiktais starptautiskais nepatentētais nosaukums
vai, ja tā nav, zinātniskais nosaukums — attiecībā uz citām
vielām. Vielas, kam nav starptautiska nepatentēta nosaukuma vai
zinātniskā nosaukuma, apraksta, izklāstot, kā un no kā tās
pagatavotas, attiecīgi papildinot ar citām svarīgām ziņām;
c) apzīmējums ar kodu E, kas tam
piešķirts un norādīts šo noteikumu 1.pielikumā, kā arī
normatīvos, kas regulē pārtikā atļautās krāsvielas, —
attiecībā uz krāsvielām;
3) lai norādītu gatavo zāļu
aktīvās(-o) vielas(-u) kvantitatīvo sastāvu, atkarībā no
attiecīgās zāļu formas precizē katras aktīvās vielas masu vai
bioloģiskās aktivitātes vienību skaitu (devas vienībā vai masas
vienībā, vai tilpuma vienībā);
4) aktīvās vielas apzīmē
kvantitatīvi savienojumu vai atvasinājumu veidā, norādot kopējo
masu un, ja nepieciešams, — molekulas aktīvās vienības(-u)
masu;
5) zālēm, kurās ir aktīvā viela,
uz kuru pirmo reizi attiecas reģistrācijas pieprasījums jebkurā
dalībvalstī, aktīvās vielas kvantitatīvo novērtējumu, kas ir sāls
vai hidrāts, sistemātiski izsaka ar molekulas aktīvās vienības
vai vienību masu. Visām vēlāk atļautajām zālēm dalībvalstīs to
kvantitatīvo sastāvu attiecībā uz vienu un to pašu aktīvo vielu
norāda vienā un tajā pašā veidā;
6) bioloģiskās aktivitātes
vienības izmanto vielām, kurām nevar definēt molekulāro formulu.
Ja Pasaules veselības organizācija ir noteikusi starptautisku
bioloģiskās aktivitātes vienību, tad lieto to. Ja nav noteikta
starptautiska bioloģiskās aktivitātes vienība, tad bioloģiskās
aktivitātes vienības izsaka tā, lai sniegtu nepārprotamu
informāciju par vielu aktivitāti, ja nepieciešams, izmantojot
Eiropas Farmakopejas vienības.
(2) Farmaceitiskā izstrāde:
1) šajā sadaļā sniedz informāciju
par zāļu izstrādes pētījumiem, kas veikti, lai noteiktu, ka
dozējuma veids, sastāvs, ražošanas process, iepakojuma
noslēgšanas sistēma, mikrobioloģiskās īpašības un lietošanas
instrukcija atbilst paredzētajam lietojumam, kas norādīts
reģistrācijas dokumentācijā;
2) šajā sadaļā aprakstītie
pētījumi atšķiras no regulārajiem kontroles testiem, kas veikti
saskaņā ar specifikācijām. Nosaka un apraksta preparāta un
procesa īpašību kritiskos parametrus, kas var ietekmēt sērijas
reproducējamību, zāļu darbību un zāļu kvalitāti. Ja nepieciešams,
par papildu datiem atsaucas uz attiecīgajām reģistrācijas
pieprasījuma dokumentācijas 4.moduļa (ziņojumi par neklīnisko
izpēti) un 5.moduļa (ziņojumi par klīnisko izpēti) nodaļām:
a) dokumentē aktīvās vielas
saderību ar palīgvielām, kā arī aktīvās vielas galvenos
fizikālķīmiskos rādītājus, kas var ietekmēt gatavā produkta
darbību vai dažādu aktīvo vielu saderību savienojumos;
b) dokumentē palīgvielu izvēli,
īpaši attiecībā uz to attiecīgajām funkcijām un
koncentrāciju;
c) sniedz gatavā produkta
izstrādes aprakstu, ņemot vērā paredzēto lietošanas veidu un
lietošanu;
d) pamato devas palielināšanu;
e) raksturo un dokumentē
parametrus, kas attiecas uz gatavā produkta darbību, ciktāl tas
attiecas uz fizikālķīmiskajām un bioloģiskajām īpašībām;
f) norāda ražošanas procesa izvēli
un optimizāciju, kā arī atšķirības starp ražošanas procesu(-iem),
ko izmanto galvenās klīniskās sērijas ražošanā, un procesu, kuru
izmanto gatavo zāļu ražošanā;
g) dokumentē iepakojuma un
noslēgšanas sistēmas piemērotību gatavā produkta glabāšanai,
pārvadāšanai un izmantošanai. Ja nepieciešams, apsver zāļu un
iepakojuma iespējamo mijiedarbību;
h) nesterilo un sterilo produktu
dozējuma mikrobioloģiskās īpašības atbilst Eiropas Farmakopejai,
un tās dokumentē atbilstoši Eiropas Farmakopejai;
i) lai sniegtu lietderīgu
informāciju par marķējumu, dokumentē gatavā produkta saderību ar
atjaunošanas šķīdinātāju(-iem) vai dozatoriem.
(3) Gatavo zāļu ražošanas
process:
1) norāda ražošanas metodes
aprakstu, ko atbilstoši šo noteikumu 18.4.apakšpunktam pievieno
zāļu reģistrācijas pieprasījumam, sagatavo tā, lai tas sniegtu
pietiekamu pārskatu par izmantoto operāciju būtību. Tajā iekļauj
vismaz:
a) norādi uz dažādajiem ražošanas
posmiem, tajā skaitā par procesa vadību un pieņemšanas
kritērijiem, lai varētu novērtēt, vai zāļu formas ražošanā
lietotais process var izraisīt nevēlamas pārmaiņas
komponentos;
b) ja ražošana ir
nepārtraukta, — pilnīgas ziņas par piesardzības pasākumiem,
kas veikti, lai nodrošinātu gatavā produkta viendabīgumu;
c) ražošanas procesa validācijai
izmantoto eksperimentālo pētījumu rezultātus, ja izmanto
nestandarta ražošanas metodi vai ja tiem ir izšķirīga nozīme
attiecībā uz zālēm;
d) sterilām zālēm —
informāciju par izmantotajiem sterilizēšanas procesiem, kā arī
aseptiskām procedūrām;
e) detalizētu informāciju par
sērijas sastāvu;
2) norāda katra ražotāja (tajā
skaitā līgumslēdzēju) un katras paredzētās ražošanas vietas vai
ražošanā un testēšanā iesaistītā objekta nosaukumu, adresi un
atbildību;
3) iekļauj datus par produkta
kontroles testiem, kurus var veikt ražošanas procesa starpposmā,
lai nodrošinātu ražošanas procesa atbilstību. Minētajiem testiem
ir svarīga nozīme, pārbaudot zāļu atbilstību formulai, ja
izņēmuma kārtā reģistrācijas pieprasījuma iesniedzējs piedāvā
pārbaudīt gatavās zāles ar analīzes metodi, kas neietver aktīvo
vielu noteikšanu (vai palīgvielu komponentu pārbaudi, kam piemēro
tādas pašas prasības kā aktīvajām vielām). Tas attiecas uz
gadījumiem, kad gatavo zāļu kvalitātes kontrole ir atkarīga no
procesa kontroles testiem ražošanas gaitā, īpaši, ja zāles definē
pēc to izgatavošanas metodes;
4) iesniedz validācijas pētījumu
aprakstu, dokumentāciju un ražošanas procesa kritisko posmu
noteikšanas rezultātus.
(4) Palīgvielu kontrole:
1) uzskaita palīgvielas(-u)
ražošanai vajadzīgos materiālus, norādot, kā katru materiālu
izmanto procesā. Sniedz informāciju par šo materiālu kvalitāti un
kontroli. Sniedz informāciju, kas pierāda, ka materiāli atbilst
paredzētās izmantošanas standartiem. Krāsvielām jāatbilst šo
noteikumu 1.pielikumā noteiktajām prasībām, kā arī normatīvu
prasībām par pārtikā atļautajām krāsvielām. Krāsvielas atbilst
normatīviem par tīrības kritērijiem, kādus piemēro pārtikas
krāsvielām;
2) detalizēti izklāsta katras
palīgvielas specifikācijas un pamato tās. Apraksta un pienācīgi
validē analītiskās procedūras;
3) īpašu uzmanību pievērš cilvēka
vai dzīvnieku izcelsmes palīgvielām. Veicot īpašus pasākumus, kas
saistīti ar dzīvnieku sūkļveida encefalopātijas pārnešanas
novēršanu, reģistrācijas pieprasījuma iesniedzējam attiecībā uz
palīgvielām jāpierāda, ka zāles tiek ražotas saskaņā ar
“Norādījumiem dzīvnieku sūkļveida encefalopātijas ierosinātāju
izplatīšanas riska samazināšanai ar zālēm” un to atjauninātajām
redakcijām, ko Eiropas Komisija publicējusi Eiropas Savienības
Oficiālajā Vēstnesī. Atbilstību minētajiem norādījumiem var
pierādīt, iesniedzot Eiropas Zāļu kvalitātes direktorātā
sertifikātu par atbilstību attiecīgajai Eiropas Farmakopejas
monogrāfijai vai arī sniedzot zinātniskus datus, kas šo
atbilstību pamato;
4) jaunas palīgvielas:
a) atbilstoši aktīvajai vielai
izmantotajai formai sniedz detalizētu informāciju par tās
palīgvielas(-u) raksturojumu, ko zālēs izmanto pirmo reizi vai ja
zāļu lietošanas veids ir jauns. Sniedz informāciju par
palīgvielas(-u) ražošanu un pārbaudēm, iekļaujot savstarpējas
norādes uz drošības datiem, kas iegūti neklīniskajā un klīniskajā
izpētē;
b) iesniedz dokumentu ar
detalizētu ķīmisko, farmaceitisko un bioloģisko informāciju. Šo
informāciju noformē tāpat kā 3.moduļa nodaļu par aktīvo(-ajām)
vielu(-ām);
c) informāciju par jaunu(-ām)
palīgvielu(-ām) var iesniegt kā atsevišķu dokumentu atbilstoši
iepriekš aprakstītajai formai. Ja reģistrācijas pieprasījuma
iesniedzējs nav jaunās palīgvielas ražotājs, minēto atsevišķo
dokumentu dara pieejamu reģistrācijas pieprasījuma iesniedzējam,
lai to iesniegtu kompetentajai iestādei (Latvijā — Valsts
zāļu aģentūrai);
d) dokumentācijas 4.modulī sniedz
papildu informāciju par toksicitātes pētījumiem ar jauno
palīgvielu;
e) informāciju par klīnisko izpēti
sniedz 5.modulī.
(5) Gatavo zāļu kontrole:
1) lai varētu pārbaudīt gatavās
zāles, zāļu sērijā ietilpst zāļu formas vienības, kas izgatavotas
no vienāda sākotnējā materiāla daudzuma un ar ko ir veiktas
vienādas ražošanas un sterilizēšanas darbības, vai, ja ražošanas
process ir nepārtraukts, visas vienības ir ražotas noteiktā
laikā;
2) ja nav attiecīga pamatojuma,
gatavās produkcijas aktīvās vielas satura maksimālā pieļaujamā
novirze ražošanas laikā nepārsniedz 5 %;
3) sniedz detalizētu informāciju
par specifikācijām (izlaide un glabāšanas laiks) un pamatojumu
par to izvēli, norāda analīzes metodes un to validāciju.
(6) Nosaka un detalizēti apraksta
references preparātus un references standartus, kas izmantoti
gatavo zāļu testēšanā, ja šī informācija nav sniegta iedaļā par
aktīvo vielu.
(7) Gatavo zāļu iepakojums un
noslēgšanas sistēma:
1) sniedz iepakojuma un
noslēgšanas sistēmas(-u) aprakstu un to specifikācijas,
identificējot arī primārā iepakojuma materiālus. Specifikācijās
iekļauj aprakstu un identifikāciju. Ja nepieciešams, iekļauj
farmakopejā neiekļautas metodes (ar validāciju);
2) par nefunkcionāliem sekundārā
iepakojuma materiāliem sniedz tikai īsu aprakstu. Par
funkcionāliem sekundārā iepakojuma materiāliem sniedz papildu
informāciju.
(8) Gatavo zāļu stabilitāte:
1) apkopo veiktos pētījumus,
izmantotos protokolus un pētījumu rezultātus;
2) atbilstošā formā detalizēti
atspoguļo stabilitātes pētījumu rezultātus, iekļaujot informāciju
par datu sagatavošanai izmantotajām analītiskajām procedūrām un
par šo procedūru validāciju. Ja nepieciešams, sniedz informāciju
par kopējo vakcīnu stabilitāti;
3) iesniedz stabilitātes protokolu
(pēc apstiprināšanas) un ar stabilitāti saistītās saistības.
IV nodaļa
4.modulis. Ziņojumi par neklīnisko izpēti
18. Forma un noformējums.
Vispārīgs 4.moduļa izklāsts ir šāds:
18.1.satura rādītājs;
18.2. ziņojumi par pētījumiem:
(1) Farmakoloģija:
1) primārā farmakodinamika;
2) sekundārā farmakodinamika;
3) drošības farmakoloģija;
4) farmakodinamiskā
mijiedarbība.
(2) Farmakokinētika:
1) analīzes metodes un validācijas
ziņojumi;
2) absorbcija;
3) izplatīšanās;
4) vielmaiņa;
5) izvadīšana no organisma;
6) farmakokinētiskā mijiedarbība
(neklīniskā izpēte);
7) citi farmakokinētiskie
pētījumi.
(3) Toksikoloģija:
1) vienas devas toksicitāte;
2) atkārtotas devas
toksicitāte;
3) genotoksicitāte:
a) in vitro;
b) in vivo (iekļaujot
toksikokinētikas novērtējumus);
4) kancerogenitāte:
a) ilglaicīgi pētījumi;
b) īslaicīgi vai vidēji ilgi
pētījumi;
c) citi pētījumi;
5) reproduktīvā toksicitāte un
ontoģenēzes toksicitāte:
a) auglība un agrīnā embrionālā
attīstība;
b) embrija (augļa) attīstība;
c) prenatālā un postnatālā
attīstība;
d) pētījumi, kuros zāļu devu dod
pēcnācējiem (dzīvnieku mazuļiem) un/vai tos papildus novērtē;
6) vietējā panesamība.
(4) Citi toksicitātes
pētījumi:
1) antigenitāte;
2) imūntoksicitāte;
3) mehānismu izpēte;
4) atkarība;
5) vielmaiņas produkti;
6) piemaisījumi;
7) pārējie;
18.3. literatūras atsauces.
19. Satura pamatprincipi un
pamatprasības. Īpašu uzmanību pievērš atsevišķajiem
elementiem.
(1) Farmakoloģiskajiem un
toksikoloģiskajiem testiem jāparāda:
1) zāļu potenciālā toksicitāte un
bīstamā vai nevēlamā toksiskā iedarbība, kas var rasties,
cilvēkiem tās lietojot saskaņā ar ieteiktajiem nosacījumiem. To
novērtē saistībā ar attiecīgo patoloģisko stāvokli;
2) zāļu farmakoloģiskās īpašības
kvalitatīvā un kvantitatīvā saistībā ar ieteikto lietojumu.
Rezultāti ir ticami un vispārēji piemērojami. Ja iespējams,
eksperimenta metodes izstrādāšanā un rezultātu novērtēšanā lieto
matemātikas un statistikas metodes. Informāciju par zāļu
ārstniecisko un toksikoloģisko potenciālu sniedz klīniskās
izpētes speciālisti.
(2) Moduļa prasības var pielāgot
atsevišķām bioloģiskām zālēm (piemēram, imunoloģiskām zālēm un no
cilvēka asins vai plazmas iegūtām zālēm), tādēļ pieprasījuma
iesniedzējs pamato izdarīto testu programmu. Izstrādājot testu
programmu, ņem vērā šādas prasības:
1) plāno testus, kam vajadzīga
produkta atkārtota lietošana, ņemot vērā antivielu iespējamo
indukciju un mijietekmi;
2) paredz reproduktīvo funkciju,
embrija, kā arī augļa un perinatālās toksicitātes, mutagēnu
potenciāla un kancerogēnu potenciāla pārbaudi. Ja par iemeslu
uzskata citus komponentus, kas nav aktīvā viela(-as), šo vielu
izņemšanas validācija var aizstāt izpēti.
(3) Pēta pirmo reizi farmācijā
lietotas palīgvielas toksikoloģiju un farmakokinētiku.
(4) Ja glabāšanas laikā iespējama
ievērojama zāļu sadalīšanās, novērtē sadalīšanās produktu
toksikoloģiju.
(5) Farmakoloģija:
1) farmakoloģijas pētījumā izmanto
divas atšķirīgas pieejas:
a) pienācīgi pēta un apraksta ar
iecerēto terapeitisko lietošanu saistītās darbības. Ja iespējams,
izmanto atzītas un validētas noteikšanas in vivo un in
vitro. Jaunas eksperimentālas metodes apraksta detalizēti,
lai tās varētu izmantot. Rezultātus izsaka kvantitatīvi
(izmantojot, piemēram, devas un iedarbības līknes, laika un
iedarbības līknes). Ja iespējams, tos salīdzina ar datiem, kas
attiecas uz vielu vai vielām, kuru terapeitiskā iedarbība ir
līdzīga;
b) reģistrācijas pieprasītājs pēta
vielas iespējamo nevēlamo farmakodinamisko iedarbību uz
fizioloģiskajām funkcijām. Šos pētījumus veic ar paredzēto un
lielāku terapeitiskās devas iedarbību. Eksperimentālās metodes,
ja vien tās nav standarta procedūras, jāraksturo tik detalizēti,
lai tās varētu izmantot, un pētnieks nosaka, vai tās ir derīgas.
Pēta reakciju iespējamās pārmaiņas, kas varētu rasties, vielu
atkārtoti lietojot;
2) farmakoloģisko pieņēmumu vai
terapeitiskās iedarbības indikāciju dēļ var būt vajadzīgi
steidzami aktīvo vielu kombināciju testi attiecībā uz zāļu
farmakodinamisko mijiedarbību:
a) farmakodinamikas pētījums
parāda mijiedarbības, kuru dēļ kombināciju ir vērts izmantot
ārstēšanā;
b) ja kombinācijas zinātnisko
pamatojumu meklē terapeitisko eksperimentu ceļā, izpētē nosaka,
vai sagaidāmo kombinācijas iedarbību var parādīt ar dzīvniekiem
un kāds ir tās nozīmīgums.
(6) Farmakokinētika:
1) farmakokinētika pēta aktīvās
vielas un tās vielmaiņas produktu darbības organismā un attiecas
uz šo vielu absorbcijas, izplatīšanās, vielmaiņas
(biotransformācijas) un izdalīšanas pētījumiem;
2) minēto dažādo posmu izpēti var
veikt galvenokārt ar fizikālām, ķīmiskām vai, iespējams,
bioloģiskām metodēm, kā arī novērojot pašas vielas faktisko
farmakodinamisko darbību;
3) informācija par izplatīšanos un
izvadīšanu ir nepieciešama, ja minētie dati ir obligāti
vajadzīgi, lai noteiktu devas cilvēkiem, kā arī attiecībā uz
ķīmijterapeitiskām vielām (piemēram, antibiotikas) un vielām,
kuru lietojums atkarīgs no to nefarmakodinamiskās iedarbības
(piemēram, vairāki diagnostikas reaģenti);
4) var veikt arī in vitro
pētījumus, salīdzinājumam ar dzīvnieku materiālu izmantojot
cilvēku materiālu (saistīšanās ar olbaltumvielām, vielmaiņa, zāļu
mijiedarbība);
5) nepieciešama farmakoloģiski
aktīvo vielu farmakokinētiskā izpēte. Veidojot jaunas
kombinācijas no zināmām vielām, kas ir pētītas saskaņā ar šiem
noteikumiem, farmakokinētiskos pētījumus drīkst neveikt, ja
toksicitātes testi un terapeitiskie eksperimenti pamato, ka tie
nav nepieciešami;
6) farmakokinētisko programmu
plāno, lai varētu salīdzināt datus par dzīvniekiem un cilvēkiem
un tos attiecīgi ekstrapolēt.
(7) Toksikoloģija:
1) vienas devas toksicitāte:
a) vienas devas toksicitātes tests
ir tādu toksisko reakciju kvalitatīvs un kvantitatīvs pētījums,
ko var izraisīt zālēs ietilpstošās aktīvās vielas vai vielu
vienreizēja lietošana tādās proporcijās un tādā fizikālķīmiskajā
stāvoklī, kādā tās faktiski sastopamas zālēs;
b) vienas devas toksiskuma testu
veic saskaņā ar attiecīgajām Eiropas Zāļu novērtēšanas aģentūras
publicētajām pamatnostādnēm;
2) atkārtotas devas
toksicitāte:
a) toksicitātes testi paredzēti
tādu fizioloģisku un/vai anatompatoloģisku pārmaiņu atklāšanai,
ko rada pārbaudāmās aktīvās vielas vai aktīvo vielu kombinācijas
atkārtota lietošana, kā arī paredzēti, lai noteiktu, kā šīs
pārmaiņas ir saistītas ar devām;
b) vēlams veikt divus
testus — īslaicīgu, kas ilgst divas līdz četras nedēļas, un
ilglaicīgu, kura ilgums ir atkarīgs no klīniskās izmantošanas
nosacījumiem. Tests apraksta iespējamo kaitīgo ietekmi, kam
klīniskajos pētījumos jāpievērš uzmanība. Testa ilgums ir
noteikts attiecīgajās Eiropas Zāļu novērtēšanas aģentūras
publicētajās pamatnostādnēs;
3) genotoksicitāte. Mutagēnā un
klastogēnā potenciāla izpētes nolūks ir atklāt izmaiņas, kādas
viela var izraisīt indivīdu vai šūnu ģenētiskajā materiālā.
Mutagēnas vielas var apdraudēt veselību, jo mutagēna iedarbība
rada risku, kas ierosina cilmes šūnu līnijas mutāciju ar
iespējamiem pārmantotiem traucējumiem, kā arī somatisku mutāciju
risku (arī tādu, kas izraisa vēzi). Visām jaunām vielām minētie
pētījumi ir obligāti;
4) kancerogenitāte. Parasti ir
vajadzīgi testi, kas atklāj kancerogēnu iedarbību:
a) pētījumus veic ar visām zālēm,
kuras pacientam klīniski jālieto ilgāku laiku (nepārtraukti vai
atkārtoti ik pa laikam);
b) pētījumus ieteicams veikt ar
zālēm, ja to iedarbība var būt kancerogēna (piemēram, preparātiem
no tās pašas klases vai ar līdzīgu struktūru) vai ja
kancerogenitāte pierādīta atkārtotas devas toksicitātes
pētījumos;
c) ar nepārprotami genotoksiskiem
savienojumiem pētījumus neveic, jo tos uzskata par starpsugu
kancerogēniem, kas apdraud cilvēkus. Ja šādas zāles ir paredzēts
lietot cilvēkiem pastāvīgi, var būt nepieciešams veikt ilglaicīgu
pētījumu, lai noteiktu agrīno tumorogēno iedarbību;
5) reproduktīvā toksicitāte un
ontoģenēzes toksicitāte:
a) ar attiecīgiem testiem pēta
iespējamo vīriešu un sieviešu reproduktīvās funkcijas
pasliktināšanos, kā arī kaitīgo ietekmi uz pēcnācējiem;
b) šajos testos ietilpst pētījumi
par iedarbību uz pieaugušu vīriešu un sieviešu reproduktīvo
funkciju, toksiskās un teratogēnās iedarbības pētījumi visās
cilvēka attīstības stadijās no ieņemšanas līdz dzimumgatavībai,
kā arī pētījumi par attiecīgo zāļu latento iedarbību, ja sieviete
tās lieto grūtniecības laikā;
c) ja šos testus neveic, to
pienācīgi pamato;
d) atkarībā no zāļu indicētā
lietojuma, ja zāles lietos arī bērni, var būt nepieciešami
papildu pētījumi par bērnu attīstību;
e) embrija, kā arī augļa
toksicitātes pētījumus parasti veic divām zīdītāju sugām, no tām
viena nav grauzēju suga. Vismaz vienai sugai veic perinatālos un
postnatālos pētījumus. Ja zināms, ka zāļu metabolisms konkrētā
sugā ir līdzīgs cilvēkam, vēlams izvēlēties šo sugu. Vēlams arī,
lai viena suga būtu tā pati, kas izmantota atkārtotas devas
toksicitātes pētījumos;
f) plānojot pētījumu, ņem vērā
uzkrāto zinātnisko pieredzi laikā, kad iesniedz reģistrācijas
pieprasījumu;
6) vietējā panesamība:
a) vietējās panesamības pētījumu
mērķis ir noteikt, vai tās ķermeņa vietas, kas var saskarties ar
zālēm, tās lietojot klīniski, panes zāles (gan aktīvās vielas,
gan palīgvielas). Testu stratēģija ir tāda, ka jebkuru mehānisko
iedarbību, kas ir zāļu lietošanai, vai tikai fizikālķīmisko
darbību nodala no toksikoloģiskās vai farmakodinamiskās
iedarbības;
b) vietējās panesamības pētījumus
veic ar preparātu, ko izstrādā lietošanai cilvēkiem, kontroles
grupas(-u) ārstēšanā izmantojot saistvielas, kā arī palīgvielas.
Ja nepieciešams, izmanto pozitīvas kontroles (references)
vielas;
c) vietējās panesamības plāns
(sugu izvēle, ilgums, lietošanas biežums un veids, devas) ir
atkarīgs no pētāmās problēmas un klīniskās lietošanas
paredzētajiem apstākļiem. Ja nepieciešams, novērš lokālus
bojājumus;
d) pētījumus ar dzīvniekiem var
aizstāt ar validētiem in vitro testiem, ja testu rezultātu
kvalitāte un drošības novērtējuma lietderīgums ir
salīdzināmi;
e) ķīmiskajām vielām, ko aplicē uz
ādas (piemēram, dermāli, rektāli, vagināli), novērtē iespējamo
jutīgumu veicinošo faktoru vismaz pēc vienas no pašlaik
pieejamajām testu sistēmām (jūrascūciņām vai lokāli
limfmezglos).
V nodaļa
5.modulis. Ziņojumi par klīnisko izpēti
20. Forma un noformējums.
Vispārīgs 5.moduļa izklāsts ir šāds:
20.1. klīnisko pētījumu ziņojumu
satura rādītājs;
20.2. visu klīnisko pētījumu
uzskaitījums tabulā;
20.3. ziņojumi par klīnisko
izpēti:
(1) Ziņojumi par
biofarmaceitiskajiem pētījumiem:
1) biopieejamības pētījumi;
2) biopieejamības un
bioekvivalences pētījumi (salīdzinoši ziņojumi);
3) in vivo-in vitro atbilstības
pētījumi;
4) bioanalīzes un analīzes
metodes.
(2) Ziņojumi par farmakokinētikas
pētījumiem, kuros izmanto cilvēka biomateriālus:
1) pētījumi, kas attiecas uz
saistīšanos ar plazmas olbaltumvielām;
2) aknu vielmaiņas un
mijiedarbības pētījumi;
3) pētījumi, kuros izmanto citus
cilvēka biomateriālus.
(3) Ziņojumi par cilvēka
farmakokinētikas pētījumiem:
1) veselu subjektu farmakokinētika
un sākotnējā panesamība;
2) pacientu farmakokinētika un
sākotnējā panesamība;
3) iekšējo faktoru
farmakokinētika;
4) ārējo faktoru
farmakokinētika;
5) populācijas
farmakokinētika.
(4) Ziņojumi par cilvēka
farmakodinamikas pētījumiem:
1) veselu subjektu
farmakodinamikas un farmakokinētikas-farmakodinamikas
pētījumi;
2) pacientu farmakodinamikas un
farmakokinētikas-farmakodinamikas pētījumi.
(5) Ziņojumi par efektivitātes un
drošības pētījumiem:
1) kontrolēta klīniskā izpēte par
indikāciju, uz kuru attiecas reģistrācijas pieprasījums;
2) nekontrolēti klīniskie
pētījumi;
3) vairāk nekā viena pētījuma datu
analīze, iekļaujot visas oficiālās integrētās analīzes,
metaanalīzes un laipošanas analīzes;
4) citi pētījumi.
(6) Ziņojumi par pieredzi pēc zāļu
izplatīšanas uzsākšanas.
20.4. literatūras atsauces.
21. Satura pamatprincipi un
pamatprasības. Īpašu uzmanību pievērš minētajiem izraudzītajiem
elementiem.
22. Klīniskie dati, ko sniedz,
ievērojot šo noteikumu 18.8.apakšpunktu, ir pietiekami, lai
sniegtu pamatotu un zinātniski derīgu atzinumu par to, vai zāles
atbilst kritērijiem, kuri reglamentē reģistrācijas piešķiršanu.
Ir svarīga prasība, lai klīniskās izpētes rezultātus darītu
zināmus (gan zālēm labvēlīgos, gan nelabvēlīgos).
23. Pirms klīniskās izpētes
vienmēr veic farmakoloģiskus un toksikoloģiskus testus ar
dzīvniekiem saskaņā ar šī pielikuma 4.moduļa prasībām. Pētnieks
iepazīstas ar farmakoloģisko un toksikoloģisko pētījumu
secinājumiem. Reģistrācijas pieprasījuma iesniedzējs iesniedz
viņam vismaz pētnieka brošūru, kurā ir visa būtiskā informācija,
kas zināma pirms klīniskās izpētes uzsākšanas (ķīmiskie,
farmaceitiskie un bioloģiskie dati, toksikoloģiskie,
farmakokinētiskie un farmakodinamiskie dati par dzīvniekiem un
iepriekšējās klīniskās izpētes dati, kā arī atbilstoši dati, kas
pamato paredzētās izpētes raksturu, mērogu un ilgumu). Pēc
pieprasījuma sniedz pilnīgus farmakoloģiskos un toksikoloģiskos
ziņojumus. Cilvēku un dzīvnieku izcelsmes materiāliem izmanto
visus iespējamos līdzekļus, lai nodrošinātu aizsardzību pret
infekcijas izraisītāju pārnešanu pirms izpētes sākuma.
24. Reģistrācijas īpašnieks
nodrošina, lai svarīgus klīniskās izpētes dokumentus (tajā skaitā
datu reģistrācijas veidlapas), kas nav subjekta slimības vēsture,
datu īpašnieki glabātu:
24.1. vismaz 15 gadus pēc izpētes
pabeigšanas vai pārtraukšanas;
24.2. vismaz divus gadus pēc
pēdējās reģistrācijas piešķiršanas Eiropas Kopienā, ja Eiropas
Kopienā vairs nav izskatāmu vai plānotu reģistrācijas
pieprasījumu;
24.3. vismaz divus gadus pēc
pētāmā preparāta klīniskās izstrādes oficiālas pārtraukšanas.
25. Subjekta slimības vēsturi
glabā saskaņā ar piemērojamajiem tiesību aktiem un normatīvajiem
aktiem tik ilgi, cik to ļauj ārstniecības iestāde, tajā skaitā
ārsta prakse.
26. Dokumentus var glabāt arī
ilgāk, ja tas noteikts normatīvajos aktos vai bijusi vienošanās
ar sponsoru. Sponsora pienākums ir informēt slimnīcu, iestādi vai
praksi, ja dokumenti vairs nav jāglabā.
27. Sponsors vai cits datu
īpašnieks glabā visu pārējo dokumentāciju, kas attiecas uz
izpēti, kamēr zāles ir atļautas. Šajā dokumentācijā ietilpst
protokols, kurā ietverts izpētes pamatojums, mērķi, statistiskā
koncepcija, metodika, nosacījumi, ar kādiem to veic un vada,
detalizēta informācija par pētāmo preparātu, lietotajām
references zālēm un placebo, darbības standartprocedūras, visi
rakstiskie atzinumi par protokolu un procedūrām, pētnieka
brošūra, datu reģistrācijas veidlapas par katru pētījuma
subjektu, nobeiguma ziņojums un audita sertifikāts(-i), ja
tāds(-i) ir. Sponsors vai turpmākais īpašnieks glabā nobeiguma
ziņojumu piecus gadus pēc tam, kad zāles vairs nav atļautas.
28. Reģistrācijas īpašnieks veic
papildu pasākumus, lai arhivētu Eiropas Kopienas pētījumu
dokumentāciju saskaņā ar normatīvajiem aktiem par zāļu klīnisko
izpēti, kā arī īstenojot detalizēti izstrādātas
pamatnostādnes.
29. Datu īpašnieka maiņu vienmēr
dokumentē.
30. Visus datus un dokumentus dara
pieejamus, ja to pieprasa attiecīgās iestādes.
31. Katras klīniskās izpētes datos
ir šāda pietiekama informācija, lai varētu veikt objektīvu
novērtējumu:
31.1. protokols, kurā ietverts
izpētes pamatojums, mērķi un statistiskā koncepcija un metodika,
nosacījumi, ar kādiem to veic un vada, kā arī detalizēta
informācija par pētāmajām zālēm;
31.2. audita sertifikāts(-i), ja
tāds(-i) ir;
31.3. pētnieku saraksts, kurā
norādīts katra pētnieka vārds, adrese, amati, kvalifikācija,
klīniskie pienākumi un izpētes vieta. Pētnieki apkopo informāciju
par katru pacientu atsevišķi, iekļaujot datu reģistrācijas
veidlapas par katru pētījuma subjektu;
31.4. nobeiguma ziņojumu paraksta
pētnieks, un, ja izpēte ir veikta vairākās vietās, — visi
pētnieki vai koordinators (atbildīgais pētnieks).
32. Minētos klīniskās izpētes
datus nosūta Valsts zāļu aģentūrai. Pēc vienošanās ar Valsts zāļu
aģentūru reģistrācijas pieprasījuma iesniedzējs var neiesniegt
daļu šīs informācijas, bet, ja Valsts zāļu aģentūra pieprasa,
nekavējoties iesniedz pilnīgu dokumentāciju. Secinājumos par
izpētē iegūtajiem pierādījumiem pētnieks izsaka atzinumu par zāļu
drošību, ja tās lieto pareizi, to panesamību, efektivitāti un
visu lietderīgo informāciju, kas attiecas uz indikācijām un
kontrindikācijām, devām un vidējo ārstēšanas ilgumu, kā arī par
īpašiem piesardzības pasākumiem, kas veicami ārstēšanas laikā, un
par pārdozēšanas klīniskajiem simptomiem. Ja izpēte ir veikta
vairākās vietās, atbildīgais pētnieks, ziņojot par izpētes
rezultātiem, secinājumos izsaka atzinumu par pētāmā preparāta
drošību un efektivitāti visu izpētes centru vārdā.
33. Apkopo klīniskos novērojumus
par katru izpēti un norāda:
(1) Ārstēto subjektu skaitu un
dzimumu.
(2) Pētāmo pacientu grupu atlasi,
sadalījumu pēc vecuma un salīdzinošos testus.
(3) To pacientu skaitu, kuri pirms
laika izslēgti no izpētes, un šādas izslēgšanas iemeslus.
(4) Ja kontrolētā izpēte veikta
saskaņā ar minētajiem nosacījumiem, norāda, vai kontroles
grupa:
1) nav ārstēta;
2) ir saņēmusi placebo;
3) ir saņēmusi citas zāles, kuru
iedarbība ir zināma;
4) ir ārstēta citādi (nevis ar
zālēm).
(5) Novēroto blakusparādību
(blakņu) biežumu.
(6) Informāciju par palielināta
riska grupas pacientiem (piemēram, veciem ļaudīm, bērniem,
sievietēm grūtniecības vai menstruāciju laikā) vai pacientiem,
kuru fizioloģiskais vai patoloģiskais stāvoklis prasa īpašu
uzmanību.
(7) Efektivitātes parametrus vai
novērtēšanas kritērijus un rezultātus (izsaka ar minētajiem
parametriem).
(8) Rezultātu statistisko
novērtējumu, ja tas paredzēts izpētes plānā, un ar to saistītos
mainīgos faktorus.
34. Pētnieks sniedz novērojumus
par:
(1) Pieraduma vai atkarības
pazīmēm vai grūtībām, ko sagādā zāļu lietošanas pārtraukšana
pacientiem.
(2) Jebkuru mijiedarbību, kas
novērota ar citām zālēm, kuras lieto vienlaikus.
(3) Kritērijiem, kas nosaka zināmu
pacientu izslēgšanu no izpētes.
(4) Nāves gadījumiem izpētes laikā
vai turpmākās uzraudzības laikā.
35. Dati, kas attiecas uz zāļu
jaunu kombināciju, ir identiski datiem, kurus sniedz par jaunām
zālēm, un tos pamato ar kombinācijas drošību un efektivitāti.
36. Ja datu nav vai tie ir daļēji,
to izskaidro. Ja izpētes gaitā iegūst negaidītus rezultātus, veic
un pārskata papildu pirmsklīniskos, toksikoloģiskos un
farmakoloģiskos pētījumus.
37. Ja zāles paredzētas ilgstošai
lietošanai, sniedz datus par visām farmakoloģiskās iedarbības
pārmaiņām pēc atkārtotas lietošanas, kā arī par devas noteikšanu
ilgstošai lietošanai.
38. Ziņojumi par
biofarmaceitiskajiem pētījumiem:
(1) Iesniedz ziņojumus par
biopieejamības pētījumiem, salīdzinošus ziņojumus par
biopieejamības un bioekvivalences pētījumiem, ziņojumus par in
vitro un in vivo atbilstības pētījumiem, kā arī par
bioanalīzes un analīzes metodēm.
(2) Biopieejamību novērtē, ja ir
jāpierāda šo noteikumu 19.punktā minētā zāļu bioekvivalence.
39. Ziņojumi par farmakokinētikas
pētījumiem, kuros izmanto cilvēka biomateriālu:
(1) Cilvēka biomateriāli ir
cilvēka izcelsmes olbaltumvielas, šūnas, audi un saistītie
materiāli, ko izmanto in vivo vai ex vivo, lai
novērtētu zāļu farmakokinētiskās īpašības.
(2) Ziņojumus sniedz par
pētījumiem, kas attiecas uz saistīšanos ar plazmas
olbaltumvielām, par aknu vielmaiņas un aktīvās vielas
mijiedarbības pētījumiem un par pētījumiem, kuros izmanto citus
cilvēka biomateriālus.
40. Ziņojumos par cilvēku
farmakokinētikas pētījumiem:
(1) Apraksta šādus
farmakokinētikas rādītājus:
1) absorbcija (ātrums un
pakāpe);
2) izplatīšanās;
3) vielmaiņa;
4) izdalīšana.
(2) Apraksta klīniski nozīmīgas
pazīmes (tajā skaitā kinētisko datu saistību ar devām), īpaši
riska grupas pacientiem, kā arī atšķirības starp pirmsklīniskajos
pētījumos izmantotajiem cilvēkiem un dzīvnieku sugām.
(3) Papildus standarta
farmakokinētikas pētījumiem ar daudziem eksemplāriem populācijas
farmakokinētikas analīzē, kuras pamatā ir retu eksemplāru atlase,
un klīniskajā izpētē var noteikt, kā iekšējie un ārējie faktori
ietekmē devas un farmakokinētikas attiecību. Sniedz ziņojumus par
farmakokinētikas un sākotnējās panesamības pētījumiem veseliem
subjektiem un pacientiem, ziņojumus par farmakokinētikas
pētījumiem, lai novērtētu iekšējo un ārējo faktoru iedarbību, un
ziņojumus par populācijas farmakokinētikas pētījumiem.
(4) Ja zāles paredzēts lietot kopā
ar citām zālēm, sniedz datus par kopējiem lietošanas testiem, lai
parādītu farmakoloģiskās iedarbības iespējamās pārmaiņas.
(5) Pēta aktīvās vielas un citu
zāļu vai vielu farmakokinētisko mijiedarbību.
41. Ziņojumi par cilvēku
farmakodinamikas pētījumiem:
(1) Parāda farmakodinamisko
darbību saistībā ar efektivitāti, ieskaitot:
1) devas un reakcijas attiecību,
kā arī tās izmaiņas laikā;
2) devas un lietošanas nosacījumu
pamatojumu;
3) ja iespējams, darbības
veidu.
(2) Apraksta farmakodinamisko
darbību, kas nav saistīta ar efektivitāti.
(3) Lai pamatotu secinājumus par
jebkuru konkrētu potenciālo terapeitisko iedarbību, nepietiek
parādīt farmakodinamisko iedarbību uz cilvēkiem.
(4) Ja zāles parasti paredzēts
lietot kopā ar citām zālēm, sniedz datus par kopējiem lietošanas
testiem, lai parādītu farmakoloģiskās iedarbības iespējamās
pārmaiņas.
(5) Pēta aktīvās vielas un citu
zāļu vai vielu farmakodinamisko mijiedarbību.
42. Ziņojumi par efektivitātes un
drošības pētījumiem:
(1) Ziņojumi par kontrolētu
klīnisko izpēti attiecībā uz indikāciju, uz kuru attiecas
reģistrācijas pieprasījums:
1) parasti klīnisko izpēti veic kā
kontrolētu klīnisko izpēti, ja iespējams, izmanto nejaušības
metodi, attiecīgi salīdzinot ar placebo un ar apstiprinātām
zālēm, kurām ir pierādīta terapeitiskā vērtība. Visus citus
plānus pamato. Kontroles grupu ārstēšana ir atšķirīga un atkarīga
arī no ētiskiem apsvērumiem un terapijas nozares. Dažos gadījumos
jauno zāļu efektivitāti var salīdzināt nevis ar placebo
iedarbību, bet ar tādu apstiprinātu zāļu efektivitāti, kurām ir
pierādīta terapeitiskā vērtība:
a) ciktāl iespējams, īpaši izpētē,
kur zāļu iedarbību nevar objektīvi izmērīt, attiecīgi rīkojas,
lai nepieļautu neobjektivitāti, — izmanto arī nejaušības
metodi un aklo metodi;
b) izpētes protokolā ir ietverts
izmantojamo statistikas metožu detalizēts apraksts, pacientu
skaits un viņu iesaistīšanas iemesli (tajā skaitā aprēķini par
izpētes apjomu), nozīmīgums un statistiskās vienības apraksts.
Pasākumus, kurus veic, lai nepieļautu neobjektivitāti, jo īpaši
nejaušības metodi, dokumentē. Ja izpētē iesaistīti daudzi
subjekti, nedrīkst uzskatīt, ka tas var aizstāt kontrolētu
izpēti;
2) datus par drošību izskata,
ņemot vērā Eiropas Komisijas publicētās pamatnostādnes, īpašu
uzmanību pievēršot gadījumiem, kuru dēļ mainīta deva vai bijis
nepieciešams vienlaikus lietot citas zāles, novēroti nopietni
nevēlami notikumi, pacients izslēgts no pētījuma vai miris.
Norāda visus palielināta riska grupas pacientus vai pacientu
grupas, īpašu uzmanību pievēršot potenciāli mazāk aizsargātiem
pacientiem, kuru skaits var būt neliels (piemēram, bērni,
grūtnieces, veci cilvēki ar vāju veselību, cilvēki, kuriem ir
izteikti vielmaiņas vai ekskrēcijas traucējumi. Apraksta drošības
novērtējuma saistību ar zāļu iespējamo lietošanu.
(2) Iesniedz ziņojumus par tādu
nekontrolētas klīniskās izpētes datu analīzi, kuri attiecas uz
vairāk nekā vienu pētījumu, un citus ziņojumus par klīnisko
izpēti.
43. Ziņojumi par pieredzi pēc zāļu
izplatīšanas uzsākšanas. Ja zāles ir atļautas trešajās valstīs,
sniedz informāciju par blakusparādībām, kas novērotas, lietojot
zāles ar tādu(-as) pašu(-as) aktīvo(-ās) vielu(-as), un, ja
iespējams, sniedz datus par lietojuma apmēriem minētajās
valstīs.
44. Datu reģistrācijas veidlapas
un atsevišķu pacientu saraksti. Datu reģistrācijas formas un
sarakstus ar atsevišķu pacientu datiem sniedz tādā pašā kārtībā
kā klīnisko pētījumu ziņojumus, indeksējot pēc pētījumiem. Tos
iesniedz saskaņā ar Eiropas Zāļu novērtēšanas aģentūras
publicētajām attiecīgajām pamatnostādnēm.
B sadaļa
Īpaša reģistrācijas dokumentācija un īpašas prasības
45. Dažām zālēm ir tādas
raksturīgas pazīmes, ka attiecīgi jāpielāgo visas šī pielikuma A
sadaļā noteiktās reģistrācijas pieprasījuma dokumentācijas
prasības. Lai ņemtu vērā šīs konkrētās situācijas, reģistrācijas
pieprasījuma iesniedzēji ievēro attiecīgu un pielāgotu
dokumentācijas noformējumu.
46. Šajā sadaļā noteiktas īpašas
prasības reģistrācijas (tirdzniecības atļaujas)
dokumentācijai:
(1) Medicīnā plaši lietotām
zālēm.
(2) Pēc būtības līdzīgām
zālēm.
(3) Īpašos gadījumos vajadzīgiem
papildu datiem.
(4) Līdzīgām bioloģiskas izcelsmes
zālēm.
(5) Zālēm ar nemainīgu
sastāvu.
(6) Dokumentācijai, kas paredzēta
reģistrācijas pieprasījumam ārkārtas apstākļos.
(7) Jauktajiem reģistrācijas
pieprasījumiem.
47. Medicīnā plaši lietotas zāles
ir zāles, kuru aktīvā(-ās) viela(-as) tiek plaši lietota(-as)
medicīnā atbilstoši šo noteikumu 19.2.apakšpunktam un kuru
efektivitāte ir atzīta un drošības līmenis ir pieņemams:
(1) Reģistrācijas pieprasījuma
iesniedzējs iesniedz šī pielikuma I daļā aprakstīto 1., 2. un 3.
moduli, bet 4. un 5.modulī detalizēti izstrādātā un zinātniskajā
bibliogrāfijā norāda neklīniskos un klīniskos rādītājus.
(2) Lai pierādītu plašo lietošanu
medicīnā, piemēro šādus īpašus noteikumus:
1) faktori, kas jāņem vērā, lai
pierādītu zāļu komponentu plašu lietošanu medicīnā, ir šādi:
a) laiks, cik ilgi viela ir
lietota;
b) vielas lietošanas kvantitatīvie
aspekti;
c) zinātniskā ieinteresētība
vielas lietošanā (atspoguļota publicētā zinātniskajā
literatūrā);
d) zinātnisko novērtējumu
saskanība;
2) atšķirīgām vielām var būt
vajadzīgi atšķirīgi laikposmi, lai noteiktu, ka tās tiek plaši
lietotas. Laiks, kas vajadzīgs, lai noteiktu, ka zāļu komponentu
plaši lieto medicīnā, nedrīkst būt mazāks par 10 gadiem, sākot no
pirmā sistemātiskā un dokumentētā minētās vielas kā zāļu
lietojuma Eiropas Kopienā;
3) reģistrācijas pieprasījuma
iesniedzēja iesniegtā dokumentācija attiecas uz visiem drošības
un efektivitātes novērtējuma aspektiem, un tā ietver attiecīgās
literatūras pārskatu vai atsauces uz to, ņemot vērā pētījumus
pirms laišanas tirgū un pēc tās un publicēto zinātnisko
literatūru par pieredzi epidemioloģiskos pētījumos (īpaši
salīdzinošos epidemioloģiskos pētījumos). Dara zināmu visu
dokumentāciju neatkarīgi no tā, vai tā ir zālēm labvēlīga vai
nelabvēlīga. Attiecībā uz noteikumiem par plašu lietošanu
medicīnā īpaši precizē, ka bibliogrāfiskajai atsaucei uz citiem
pierādījumu avotiem (piemēram, pētījumi pēc laišanas tirgū,
epidemioloģiskie pētījumi), nevis tikai uz datiem, kas saistīti
ar testiem un izpēti, var būt derīgs preparāta drošības un
efektivitātes apliecinājums, ja reģistrācijas pieprasījumā šo
informācijas avotu izmantošana ir pietiekami paskaidrota un
pamatota;
4) īpašu uzmanību pievērš visai
trūkstošajai informācijai un sniedz pamatojumu, kāpēc var
uzskatīt, ka drošības un efektivitātes pakāpe ir pieņemama, kaut
arī trūkst dažu pētījumu;
5) neklīniskās un klīniskās
izpētes pārskatos paskaidro visu to iesniegto datu nozīmi, kas
attiecas uz produktu, kurš atšķiras no produkta, kas paredzēts
izplatīšanai. Novērtē, vai pētāmās zāles var uzskatīt par
līdzīgām zālēm, par ko iesniegts reģistrācijas pieprasījums, kaut
arī starp tām pastāv atšķirības;
6) pieredzei, kas gūta pēc tam,
kad laistas tirgū citas zāles, kuras satur tos pašus komponentus,
ir īpaša nozīme, un reģistrācijas pieprasījuma iesniedzēji īpaši
izskata šos jautājumus.
48. Attiecībā uz būtībā līdzīgām
zālēm:
(1) Reģistrācijas pieprasījumos,
kuri pamatoti ar šo noteikumu 19.1.apakšpunktu (būtībā līdzīgs
produkts), iekļauj šī pielikuma A sadaļas 1., 2. un 3.modulī
aprakstītos datus, ja oriģinālo zāļu reģistrācijas īpašnieks ir
piekritis tam, ka reģistrācijas pieprasījuma iesniedzējs izmanto
savstarpējas norādes uz oriģinālo zāļu reģistrācijas īpašnieka
iesniegto 4. un 5.moduļa saturu.
(2) Reģistrācijas pieprasījumos,
kuri pamatoti ar šo noteikumu 19.4.apakšpunktu (būtībā līdzīgs
produkts- generic zāles), iekļauj šī pielikuma A sadaļas 1., 2.
un 3.modulī aprakstītos datus un dokumentus, kas parāda
biopieejamību un bioekvivalenci salīdzinājumā ar oriģinālajām
zālēm, ja oriģinālās zāles nav bioloģiskas izcelsmes zāles (IV
nodaļa).
(3) Attiecībā uz šiem produktiem
neklīniskās (klīniskās) izpētes pārskatos (kopsavilkumos) galveno
uzmanību pievērš šādiem elementiem:
1) pamatojumam, kādēļ tiek
pieprasīta ļoti liela līdzība;
2) kopsavilkumam par
piemaisījumiem, kas atrodami tās (to) aktīvās(-o) vielas(-u), kā
arī gatavo zāļu sērijās (ja nepieciešams, sadalīšanās produkti,
kas rodas glabāšanas laikā), kuras iecerēts izmantot tirgū
laižamajā produktā, un šo piemaisījumu novērtējumam;
3) bioekvivalences pētījumu
novērtējumam vai pamatojumam, lai neveiktu pētījumus attiecībā uz
pamatnostādnēm par biopieejamības un bioekvivalences izpēti;
4) atjauninātai publicētai
literatūrai, kas attiecas uz vielu un konkrēto reģistrācijas
pieprasījumu. Šādā nolūkā var anotēt rakstus no profesionālajiem
žurnāliem;
5) zāļu aprakstā jebkuru
apgalvojumu, kas nav zināms vai ko nevar secināt no zāļu
raksturīgajām pazīmēm vai to terapeitiskās grupas, aplūko
neklīniskās (klīniskās) izpētes pārskatos (kopsavilkumos) un
pamato ar literatūras publikācijām, kā arī papildu
pētījumiem;
6) ja reģistrācijas pieprasījuma
iesniedzēja pieprasījums attiecas uz ļoti lielu līdzību, tam
jāsniedz papildu dati, lai pierādītu dažādu atļautās aktīvās
vielas sāļu, esteru vai atvasinājumu drošības un efektivitātes
īpašību līdzvērtīgumu.
49. Īpašās situācijas, kad
vajadzīgi papildu dati:
(1) Ja būtībā līdzīgu zāļu aktīvā
viela satur vienu un to pašu ārstniecisko sastāvdaļu, ko
oriģinālais atļautais produkts, kas saistīts ar atšķirīgu sāls,
kā arī estera kompleksu, kā arī atvasinājumu, pierāda, ka
sastāvdaļu farmakokinētikā, farmakodinamikā, kā arī toksicitātē
nav izmaiņu, kas varētu mainīt drošības, kā arī efektivitātes
raksturojumu. Ja izmaiņas ir, šo savienojumu uzskata par jaunu
aktīvo vielu.
(2) Ja zāles paredzētas citādai
terapeitiskai lietošanai vai zāļu forma ir citāda, vai zāles
paredzēts lietot citādos veidos un devās, vai zāļu devas ir
atšķirīgas, sniedz attiecīgu toksikoloģisko un farmakoloģisko
testu, kā arī klīniskās izpētes rezultātus.
50. Līdzīgas bioloģiskas izcelsmes
zāles:
(1) Attiecībā uz bioloģiskas
izcelsmes zālēm šo noteikumu 19.4.apakšpunkta nosacījumi var būt
nepietiekami. Ja ar informāciju, kas vajadzīga par būtībā
līdzīgiem produktiem (generic zāles), nevar pierādīt divu
bioloģiskas izcelsmes zāļu līdzīgo būtību, sniedz papildu datus,
jo īpaši toksikoloģisko un klīnisko raksturojumu.
(2) Ja neatkarīgs reģistrācijas
pieprasījuma iesniedzējs pēc datu aizsardzības perioda beigām
iesniedz reģistrācijas pieprasījumu šī pielikuma A sadaļas
17.punktā noteiktajām bioloģiskas izcelsmes zālēm, par kurām ir
dota atsauce uz oriģinālajām zālēm, kam Eiropas Kopienā ir
piešķirta zāļu reģistrācijas apliecība, veic šādas darbības:
1) iesniedzamo informāciju par 1.,
2. un 3.moduli (farmaceitiskie, ķīmiskie un bioloģiskie dati)
papildina ar datiem par bioekvivalenci un biopieejamību. Papildu
datu veidu un apjomu (toksikoloģiskiem un citiem neklīniskajiem
datiem, kā arī attiecīgas klīniskās izpētes datiem) nosaka katrā
gadījumā atsevišķi saskaņā ar attiecīgajām pamatnostādnēm;
2) bioloģiskas izcelsmes zāļu
dažādības dēļ kompetentā iestāde (Latvijā — Valsts zāļu
aģentūra) pieprasa noteiktus 4. un 5.modulī paredzētus pētījumus,
ņemot vērā katru atsevišķu zāļu īpašās raksturīgās pazīmes.
(3) Vispārīgie principi, ņemot
vērā konkrēto bioloģiskas izcelsmes zāļu raksturīgās pazīmes, ir
norādīti Eiropas Zāļu novērtēšanas aģentūras publicētajās
pamatnostādnēs. Ja sākotnēji atļautajām zālēm ir vairāk par vienu
indikāciju, uz kuru attiecas reģistrācijas pieprasījums,
līdzīgumu pamato vai, ja nepieciešams, pierāda atsevišķi katrai
indikācijai, uz kuru attiecas reģistrācijas pieprasījums.
51. Zāles ar nemainīgu
sastāvu:
(1) Reģistrācijas pieprasījumi,
kuri pamatoti ar šo noteikumu 21.punktu, attiecas uz tādām jaunām
zālēm, kas pagatavotas no vismaz divām aktīvajām vielām un kas
nav iepriekš atļautas kā zāles ar nemainīgu sastāvu.
(2) Attiecībā uz šādiem
reģistrācijas pieprasījumiem iesniedz pilnīgu dokumentāciju (1.,
2., 3., 4. un 5.modulis) par zālēm ar nemainīgu sastāvu. Ja
nepieciešams, sniedz informāciju par ražošanas vietu un iespējamo
slimības ierosinātāju drošības novērtējumu.
52. Dokumentācija reģistrācijas
pieprasījumiem ārkārtas apstākļos:
(1) Reģistrāciju var piešķirt,
ievērojot īpašas saistības, ja reģistrācijas pieprasījuma
iesniedzējs atbilstoši šo noteikumu 135.4.apakšpunktā norādītajām
prasībām var pierādīt, ka nespēj sniegt visaptverošus datus par
efektivitāti un drošību pareizas lietošanas apstākļos, jo:
1) indikācijas, kam attiecīgās
zāles ir paredzētas, sastopamas tik reti, ka nevar gaidīt, lai
reģistrācijas pieprasījuma iesniedzējs sniegtu visaptverošus
pierādījumus;
2) visaptverošu informāciju nevar
sniegt līdz šim uzkrātās zinātniskās pieredzes dēļ;
3) šādas informācijas vākšana būtu
pretrunā vispārpieņemtiem medicīnas ētikas principiem,
reģistrāciju var piešķirt, ievērojot noteiktas īpašas
saistības.
(2) Šajā gadījumā reģistrāciju
piešķir, ievērojot šādus nosacījumus:
1) Valsts zāļu aģentūras
noteiktajā laikā reģistrācijas pieprasījuma iesniedzējs pabeidz
noteikto pētījumu programmu, kuras rezultāti būs atkārtota
ieguvumu un riska novērtējuma pamatā;
2) attiecīgās zāles drīkst pārdot
tikai ar ārsta recepti, dažos gadījumos to var lietot tikai
stingrā medicīniskā uzraudzībā, ja nepieciešams, slimnīcā.
Radiofarmaceitisku preparātu uzraudzību veic pilnvarota
persona;
3) lietošanas instrukcija un
medicīniskā informācija vērš ārsta uzmanību uz to, ka par
attiecīgajām zālēm pieejamie dati vēl nav pietiekami.
53. Jaukti reģistrācijas
pieprasījumi ir reģistrācijas pieprasījumu dokumentācija, ja 4.
un 5.modulis sastāv no ziņojumiem par ierobežotu neklīnisko un
klīnisko izpēti, ko veicis reģistrācijas pieprasījuma
iesniedzējs, kā arī no bibliogrāfiskām norādēm. Visi pārējie
moduļi atbilst šī pielikuma A daļā aprakstītajai struktūrai.
Valsts zāļu aģentūra akceptē reģistrācijas pieprasījuma formu,
katru gadījumu izskatot atsevišķi.
C sadaļa
Specifiskas zāles
54. Šajā sadaļā noteiktas īpašas
prasības attiecībā uz noteiktu zāļu īpašībām:
(1) Bioloģiskas izcelsmes zālēm
(no cilvēka asinīm un plazmas iegūtas zāles un vakcīnas).
(2) Radiofarmaceitiskiem
produktiem.
(3) Radiofarmaceitiskiem
prekursoriem (prekursors — visi pārējie radionuklīdi, ko
ražo citas vielas radioaktīvajai iezīmēšanai pirms tās lietošanas
radioaktīvai iezīmēšanai).
(4) Homeopātiskām zālēm.
(5) Augu izcelsmes zālēm.
(6) Zālēm reti sastopamu slimību
ārstēšanai.
55. No cilvēka asinīm un plazmas
iegūtās zāles. Neņemot vērā 3.moduļa nosacījumus, minēto zāļu
dokumentācijas daļā “Informācija par izejvielām un neapstrādātiem
materiāliem” informāciju par izejvielām var aizstāt ar plazmas
pamatdatni, kas ir apstiprināta, pamatojoties uz šādiem
principiem:
(1) Šajā pielikumā:
1) plazmas pamatlieta ir no
reģistrācijas pieprasījuma dokumentācijas atsevišķa
dokumentācija, kurā sniegta detalizēta informācija par
raksturīgajām pazīmēm cilvēka plazmai, kas kā izejviela un kā
neapstrādāts materiāls izmantots apakšfrakciju un starpfrakciju
palīgvielas(-u) un aktīvās(-o) vielas(-u) komponentu ražošanā un
ir daļa no zālēm vai normatīvajos aktos noteiktajām medicīnas
ierīcēm, kurās izmanto stabilus cilvēka asins vai cilvēka asins
plazmas atvasinājumus;
2) katrs cilvēka plazmas
frakcionēšanas un apstrādes centrs vai iestāde sagatavo un
atjaunina plazmas pamatlietā minēto attiecīgās detalizētās
informācijas kopumu;
3) plazmas pamatlietu Eiropas Zāļu
novērtēšanas aģentūrā vai kompetentajā iestādē (Latvijā -Valsts
zāļu aģentūrai) iesniedz reģistrācijas pieprasījuma iesniedzējs
vai reģistrācijas īpašnieks. Ja reģistrācijas pieprasījuma
iesniedzējs vai reģistrācijas īpašnieks nav plazmas pamatlietas
turētājs, plazmas pamatlietu dara pieejamu reģistrācijas
pieprasījuma iesniedzējam vai reģistrācijas īpašniekam, lai
varētu to iesniegt kompetentajai iestādei (Latvijā — Valsts
zāļu aģentūrai). Reģistrācijas pieprasījuma iesniedzējs vai
reģistrācijas īpašnieks uzņemas atbildību par zālēm;
4) Valsts zāļu aģentūra, kas
novērtē reģistrācijas pieprasījumu, nepieņem lēmumu, līdz Eiropas
Zāļu novērtēšanas aģentūra izdod sertifikātu;
5) reģistrācijas pieprasījuma
dokumentācijā, kas attiecas uz komponentu, kurš iegūs no cilvēka
plazmas, vienmēr atsaucas uz plazmas pamatlietu par plazmu, kas
izmantota kā izejviela vai neapstrādāts materiāls.
(2) Saturs. Plazmas pamatlietā
iekļauj šādu informāciju par plazmu, kas izmantota kā izejviela
vai neapstrādāts materiāls:
1) plazmas izcelsme:
a) informācija par centriem vai
iestādēm, kur savāc asinis un plazmu, iekļaujot arī datus par
inspekcijām un apstiprinājumu, kā arī epidemioloģiskie dati par
infekcijām, ko pārnēsā ar asinīm;
b) informācija par centriem vai
iestādēm, kur veic donoru materiālu un plazmas fondu testēšanu,
iekļaujot arī datus par inspekcijām un apstiprinājumu;
c) asins un plazmas donoru
izvēles, kā arī izslēgšanas kritēriji;
d) izmantotā sistēma, kas
nodrošina, ka var izsekot katra donora nodotā materiāla ceļu no
asins un plazmas savākšanas līdz gatavajam produktam un
otrādi;
2) plazmas kvalitāte un
drošība:
a) atbilstība Eiropas Farmakopejas
monogrāfijām;
b) asins un plazmas materiālu un
fondu testēšana attiecībā uz infekcijas izraisītājiem, arī
informācija par testa metodēm un attiecībā uz plazmas fondu
(validācijas dati par izmantotajiem testiem);
c) asins un plazmas savākšanai
lietoto maisiņu tehniskais raksturojums, arī informācija par
lietoto antikoagulanta šķīdumu;
d) plazmas glabāšanas un
transportēšanas apstākļi;
e) veiktās inventarizācijas
procedūras un/vai karantīnas periods;
f) plazmas fonda raksturojums;
3) izmantotā sistēma, kas nosaka
no plazmas iegūto zāļu ražotāja, plazmas frakcionētāja un
apstrādātāja, no vienas puses, un asins un plazmas savākšanas un
testēšanas centru vai iestāžu, no otras puses, mijiedarbības
nosacījumus un specifikācijas, par ko panākta vienošanās.
Papildus tam plazmas pamatlietā iekļauj to zāļu sarakstu, uz
kurām attiecas plazmas pamatlieta, neatkarīgi no tā, vai zāles ir
reģistrētas vai arī atrodas reģistrācijas procesā (arī par zālēm
saistībā ar labas klīniskās prakses ieviešanu klīniskajā izpētē
ar cilvēkiem paredzētajām zālēm).
(3) Novērtēšana un
sertificēšana:
1) par vēl neatļautām zālēm
reģistrācijas iesniedzējs kompetentajai iestādei (Latvijā —
Valsts zāļu aģentūrai) iesniedz pilnīgu dokumentāciju un
atsevišķu plazmas pamatlietu, ja tāda nav iesniegta;
2) plazmas pamatlietu zinātniski
un tehniski novērtē Eiropas Zāļu novērtēšanas aģentūra. Ja
novērtējums ir pozitīvs, par plazmas pamatlietu izdod
sertifikātu, kas apliecina, ka tā atbilst Eiropas Kopienas
tiesību aktiem, kā arī novērtējuma ziņojumu. Izdotais sertifikāts
ir derīgs visā Eiropas Kopienā;
3) plazmas pamatlietu atjaunina un
katru gadu sertificē atkārtoti;
4) veiktās izmaiņas plazmas
pamatlietas noteikumos novērtē atbilstoši procedūrai, kas
noteikta Eiropas Komisijas 2003.gada 3.jūnija Regulā (EK)
Nr.1085/2003 par izmaiņu izskatīšanu cilvēkiem paredzētu zāļu un
veterināro zāļu tirdzniecības atļaujas nosacījumos, uz ko
attiecas Regula, ar ko nosaka Kopienas procedūru tam, kā
apstiprināt un pārraudzīt zāles, kuras paredzētas izmantošanai
cilvēkiem un veterinārijā, un ar ko nodibina Eiropas Zāļu
novērtēšanas aģentūru (turpmāk — Komisijas regula
Nr.1085/2003);
5) Valsts zāļu aģentūra ņem vērā
attiecīgo zāļu plazmas pamatlietas sertifikāciju, atkārtotu
sertifikāciju vai izmaiņas;
6) ja plazmas pamatlieta attiecas
tikai uz tām no asins un plazmas iegūtajām zālēm, kuru
reģistrācija ir derīga tikai Latvijas Republikā, minētās plazmas
pamatlietas zinātnisko un tehnisko novērtējumu veic Valsts zāļu
aģentūra.
56. Vakcīnas. Ja izmanto vakcīnas
antigēnu pamatlietas sistēmu attiecībā uz cilvēkiem paredzētām
vakcīnām, neņemot vērā 3.moduļa nosacījumus, piemēro šādas
prasības — vakcīnas reģistrācijas pieprasījuma
dokumentācijā, kas nav gripas vakcīna, iekļauj katra vakcīnas
antigēna pamatlietu, kura ir minētās vakcīnas aktīvā viela:
(1) Principi:
1) vakcīnas antigēnu pamatlieta ir
atsevišķa vakcīnas reģistrācijas pieprasījuma dokumentācijas
daļa, kurā ir ietverta visa attiecīgā informācija par to aktīvo
vielu bioloģiskajām, farmaceitiskajām un ķīmiskajām īpašībām, kas
ir šo zāļu sastāvdaļa. Atsevišķā daļa var attiekties uz vienu vai
vairākām monovakcīnām, kā arī uz kombinētajām vakcīnām, ko
iesniedz viens un tas pats reģistrācijas pieprasījuma iesniedzējs
vai reģistrācijas īpašnieks;
2) vakcīna var saturēt vienu vai
vairākus atšķirīgus vakcīnas antigēnus. Vakcīnā ir tikpat
aktīvās(-o) vielas(-u), cik vakcīnas antigēna(-u);
3) kombinēta vakcīna satur vismaz
divus atšķirīgus vakcīnas antigēnus, kas aizsargā no vienas vai
vairākām infekcijas slimībām;
4) monovakcīna satur vienu
vakcīnas antigēnu, kas aizsargā no vienas infekcijas
slimības.
(2) Saturs. Vakcīnas antigēnu
pamatlietā ietver šādu informāciju no 3.moduļa attiecīgās daļas
(aktīvā viela) par kvalitātes datiem, kā noteikts šī pielikuma A
sadaļā. Attiecībā uz aktīvo vielu norāda:
1) vispārīgu informāciju, tajā
skaitā atbilstība attiecīgajai(-ajām) Eiropas Farmakopejas
monogrāfijai(-ām);
2) informāciju par aktīvās vielas
ražošanu (ražošanas procesa apraksts, informācija par izejvielām
un neapstrādātiem materiāliem, īpašiem pasākumiem saistībā ar
TSE, iespējamo slimības ierosinātāju drošības novērtējums, kā arī
iekārtas un aprīkojums);
3) aktīvās vielas
raksturojumu;
4) aktīvās vielas kvalitātes
kontroli;
5) references standartus un
materiālus;
6) aktīvās vielas iepakojumu un
noslēgšanas sistēmu;
7) aktīvās vielas stabilitāti.
(3) Novērtēšanā un sertificēšanā
ņem vērā, ka:
1) par vakcīnām, kas satur jaunu
vakcīnas antigēnu, reģistrācijas pieprasījuma iesniedzējs
kompetentai iestādei (Latvijā — Valsts zāļu aģentūrai)
iesniedz pilnīgu reģistrācijas pieprasījuma dokumentāciju,
iekļaujot visas vakcīnas antigēnu pamatlietas par katru atsevišķu
vakcīnas antigēnu, kas ir jaunās vakcīnas daļa, ja šāda atsevišķa
vakcīnas antigēna pamatlieta nav iesniegta. Eiropas Zāļu
novērtēšanas aģentūra veic visu vakcīnas antigēnu pamatlietu
zinātnisko un tehnisko novērtējumu. Ja novērtējums ir pozitīvs,
par katru vakcīnas antigēnu pamatlietu izdod sertifikātu, kas
apliecina, ka tā atbilst Eiropas Kopienas tiesību aktiem, kā arī
sniedz novērtējuma ziņojumu. Sertifikāts ir derīgs visā Eiropas
Kopienā;
2) minētie nosacījumi attiecas arī
uz visām vakcīnām, kas sastāv no jaunas vakcīnas antigēnu
kombinācijas, neatkarīgi no tā, vai viens vai vairāki vakcīnas
antigēni ir Eiropas Kopienā jau atļautu vakcīnu daļa;
3) Eiropas Zāļu novērtēšanas
aģentūra zinātniski un tehniski novērtē satura izmaiņas, kas
veiktas Eiropas Kopienā atļautas vakcīnas antigēnu pamatlietā
saskaņā ar Komisijas regulā Nr.1085/2003 noteikto procedūru. Ja
novērtējums ir pozitīvs, Eiropas Zāļu novērtēšanas aģentūra par
vakcīnas antigēnu pamatlietu izdod sertifikātu, kas apliecina, ka
tā atbilst Eiropas Kopienas tiesību aktiem. Izdotais sertifikāts
ir derīgs visā Eiropas Kopienā;
4) ja vakcīnas antigēnu pamatlieta
attiecas tikai uz vakcīnu, kam reģistrācija nav vai netiks
piešķirta saskaņā ar Eiropas Kopienas procedūru, tad, neņemot
vērā “1”, “2” un “3” punktā minētos nosacījumus, ja atļautajā
vakcīnā ietilpst vakcīnas antigēni, kas nav novērtēti saskaņā ar
Eiropas Kopienas procedūru, minētās vakcīnas antigēnu pamatlietas
un no tās izrietošo izmaiņu zinātnisko un tehnisko novērtējumu
veic Valsts zāļu aģentūra;
5) Valsts zāļu aģentūra ņem vērā
attiecīgo zāļu vakcīnas antigēnu pamatlietas sertifikāciju,
atkārtotu sertifikāciju vai izmaiņas.
57. Radiofarmaceitisko produktu
reģistrācijas pieprasījumiem, kuri pamatoti ar šo noteikumu 2.3.
un 18.16.apakšpunktā minētajiem nosacījumiem, iesniedz pilnīgu
dokumentāciju, kurā iekļauj šādu īpašu informāciju:
(1) 3.modulis:
1) attiecībā uz radiofarmaceitisko
komplektu, kam ražotājs pēc piegādes veic radioaktīvo iezīmēšanu,
aktīvo vielu uzskata par preparāta daļu, kurai paredzēts nest vai
saistīt radionuklīdu. Radiofarmaceitisko komplektu ražošanas
metodes aprakstā iekļauj informāciju par komplekta ražošanu un
informāciju par ieteicamo galīgo apstrādi, kurā iegūst
radiofarmaceitisko produktu. Vajadzīgās radionuklīda
specifikācijas, ja nepieciešams, apraksta saskaņā ar Eiropas
Farmakopejas vispārīgo monogrāfiju vai speciālām monogrāfijām.
Apraksta visus savienojumus, kas ir svarīgi radioaktīvajā
iezīmēšanā. Apraksta arī radioaktīvi iezīmētā savienojuma
struktūru. Attiecībā uz radionuklīdiem apraksta attiecīgās
kodolreakcijas. Ģeneratorā par aktīvajām vielām uzskata gan
primāros, gan sekundāros radionuklīdus;
2) norāda radionuklīda īpašības,
izotopa identitāti, iespējamos piemaisījumus, nesēju, lietošanu
un konkrēto iedarbību;
3) izejvielas ietver apstarošanas
mērķa materiālus;
4) sniedz apsvērumus par ķīmisko,
kā arī radioķīmisko tīrību un tās saistību ar bioloģisko
sadalījumu;
5) apraksta radionuklīda tīrību,
radioķīmisko tīrību un īpašo aktivitāti;
6) attiecībā uz ģeneratoriem
norāda informāciju par primāro un sekundāro radionuklīdu testiem.
Attiecībā uz ģeratoru eluātiem sniedz informāciju par primāro
radionuklīdu un citu ģeneratora sistēmas komponentu testiem;
7) prasību izteikt aktīvo vielu
saturu ar aktīvo vienību masu attiecina tikai uz
radiofarmaceitisko preparātu iepakojumiem. Attiecībā uz
radionuklīdiem radioaktivitāti izsaka bekerelos konkrētā dienā
un, ja nepieciešams, laikā, norādot laika zonu. Norāda radiācijas
veidu;
8) attiecībā uz komplektiem gatavo
zāļu specifikācijās iekļauj zāļu iedarbības testus pēc
radioaktīvās iezīmēšanas. Iekļauj atbilstošas radioaktīvi
iezīmētā savienojuma radioķīmiskās un radionuklīda tīrības
pārbaudes. Identificē un nosaka visus materiālus, kas ir svarīgi
radioaktīvajā iezīmēšanā;
9) sniedz informāciju par
radionuklīdu ģeneratoru, radionuklīdu komplektiem un radioaktīvi
iezīmētu produktu stabilitāti. Dokumentē pudelītēs iepakotu
radiofarmaceitisko produktu vairāku devu stabilitāti lietošanas
laikā.
(2) 4.modulis. Toksicitāti vēlams
saistīt ar radiācijas devu. Diagnozē ir radiofarmaceitisko
produktu lietošanas sekas, terapijā tā ir vēlamā īpašība.
Radiofarmaceitisko preparātu drošības un efektivitātes
novērtējumā pievēršas prasībām attiecībā uz zālēm un radiācijas
dozimetrijas aspektiem. Dokumentē radiācijas iedarbību uz
orgāniem un audiem. Absorbētās radiācijas devas novērtējumu
aprēķina saskaņā ar konkrētu starptautiski atzītu sistēmu
atbilstoši konkrētajam lietošanas veidam.
(3) 5.modulis. Ja nepieciešams,
sniedz klīniskās izpētes rezultātus, kas citādi pamatoti
klīniskās izpētes pārskatos.
58. Attiecībā uz
radiofarmaceitiskiem prekursoriem radioaktīvai iezīmēšanai, ja
tikai radioaktīvajai iezīmēšanai paredzēta prekursora galvenais
mērķis ir sniegt informāciju par iespējamām vājas radioaktīvās
iezīmēšanas iedarbības sekām vai in vivo radioaktīvi iezīmēta
konjugāta disociāciju (t.i., par jautājumiem, kas saistīti ar
iedarbību, ko pacientam rada brīvie radionuklīdi), sniedz arī
attiecīgo informāciju par arodapdraudējumu — radiācijas
iedarbību uz slimnīcas personālu un vidi. Minētajā gadījumā
norāda šādu informāciju:
(1) 3.moduļa noteikumi atbilstoši
šī pielikuma 57.punkta pirmās daļas nosacījumiem attiecas uz
radiofarmaceitisko prekursoru reģistrāciju.
(2) 4.modulis sniedz pētījumu
rezultātus par vienreizējas devas un atkārtotas devas
toksicitāti, kas veikti saskaņā ar normatīvajiem aktiem par labu
laboratorijas praksi, ja vien nav pamata rīkoties citādi.
Minētajā gadījumā mutagenitātes pētījumi ar radionuklīdu nav
uzskatāmi par derīgiem. Sniedz informāciju par attiecīgā “aukstā”
nuklīda ķīmisko toksicitāti un dispozīciju.
(3) 5.modulis. Prekursora
klīniskās izpētes rezultātā iegūtā informācija nav uzskatāma par
nozīmīgu attiecībā uz konkrēto prekursoru, kas paredzēts tikai
radioaktīvajai iezīmēšanai. Sniedz informāciju, kas pierāda
radiofarmaceitiskā prekursora lietderību, ja tas ir piesaistīts
attiecīgajām nesējmolekulām.
59. Šajā iedaļā ietverti īpaši
noteikumi par 3. un 4.moduļa piemērošanu homeopātiskajām
zālēm:
(1) 3.moduļa noteikumi attiecas uz
saskaņā ar šo noteikumu 21.punktu iesniegtajiem dokumentiem
homeopātisko zāļu vienkāršotajai reģistrācijas procedūrai, kā arī
uz dokumentiem citu homeopātisko zāļu reģistrācijai ar turpmākām
izmaiņām:
1) terminoloģijā. Reģistrācijas
pieprasījuma dokumentācijā aprakstītais homeopātiskās izejvielas
nosaukums latīņu valodā saskan ar Eiropas Farmakopejā lietoto
nosaukumu latīņu valodā vai, ja tur tāda nav, — dalībvalsts
oficiālajā farmakopejā. Ja nepieciešams, sniedz katrā dalībvalstī
izmantoto(-os) tradicionālo(-os) nosaukumu(-us);
2) izejmateriālu kontrolē:
a) kopā ar reģistrācijas
pieprasījumu iesniegtos datus un dokumentus par izejvielām (par
visiem materiāliem, tajā skaitā par neapstrādātiem materiāliem un
starpsavienojumiem līdz pat galīgajam atšķaidījumam, kas
jāiekļauj gatavajās zālēs) papildina ar datiem par homeopātisko
izejvielu;
b) vispārīgās kvalitātes prasības
attiecas uz visām izejvielām un neapstrādātiem materiāliem, kā
arī uz ražošanas procesa starpposmiem līdz pat galīgajam
atšķaidījumam, kas iekļaujams gatavajās zālēs. Pēc iespējas
nosaka iekļaujamo galīgo atšķaidījumu, ja sastāvā ir toksiski
komponenti un ja kvalitāti nevar kontrolēt augstās atšķaidījuma
pakāpes dēļ. Pilnībā apraksta zāļu ražošanas procesu no
izejvielām līdz galīgajam atšķaidījumam;
c) ja ir iekļauti atšķaidījumi,
atšķaidīšanu veic saskaņā ar homeopātiskās ražošanas metodēm, kas
noteiktas Eiropas Farmakopejas attiecīgajā monogrāfijā vai, ja
tur tādu nav, — dalībvalsts oficiālajā farmakopejā;
3) gatavo zāļu kontroles
testos:
a) vispārīgās kvalitātes prasības
attiecas uz gatavajām homeopātiskajām zālēm. Jebkuru izņēmumu
reģistrācijas pieprasījuma iesniedzējs pienācīgi pamato;
b) identificē un nosaka visus
toksikoloģiski nozīmīgos komponentus. Ja var pamatot, ka visu
toksikoloģiski nozīmīgo komponentu identificēšana un/vai
noteikšana nav iespējama (piemēram, atšķaidījuma pakāpes dēļ),
kvalitāti pierāda ar ražošanas un atšķaidīšanas procesa pilnīgu
validāciju;
4) stabilitātes testi pierāda
gatavo zāļu stabilitāti. Homeopātisko izejvielu stabilitātes dati
parasti ir attiecināmi uz atšķaidījumiem, kā arī uz saberzto
masu, ko no tām iegūst. Ja aktīvās vielas identifikācija vai
noteikšana nav iespējama atšķaidījuma pakāpes dēļ, var ņemt vērā
zāļu formas stabilitātes datus.
(2) 4.moduļa nosacījumi attiecas
uz šo noteikumu 3.punktā minēto homeopātisko zāļu vienkāršotu
reģistrāciju ar papildu specifikācijām. Trūkstošo informāciju
pamato (piemēram, pamato, kādēļ drošības līmeņa pierādījums ir
pieņemams, ja nav dažu pētījumu).
60. Attiecībā uz augu izcelsmes
zālēm zāļu reģistrācijas pieprasījumos sniedz pilnīgu
dokumentāciju, kurā iekļauj šādas konkrētas ziņas:
60.1. 3.moduļa nosacījumi, tajā
skaitā atbilstība Eiropas Farmakopejas monogrāfijai(-ām),
attiecas uz augu izcelsmes zāļu reģistrāciju. Ņem vērā uzkrāto
zinātnisko pieredzi laikā, kad iesniegts reģistrācijas
pieprasījums;
60.2. izskata augu izcelsmes zāļu
specifiskus aspektus:
(1) Augu izcelsmes vielas un augu
izcelsmes preparāti:
1) sadaļā “termini” augu izcelsmes
vielas un preparāti ir pielīdzināti terminiem, kas definēti
Eiropas Farmakopejā;
2) attiecībā uz augu izcelsmes
vielas nomenklatūru norāda auga binominālo zinātnisko nosaukumu
(ģints, suga, šķirne un autors) un ķīmisko tipu (attiecīgā
gadījumā), augu daļas, augu izcelsmes vielas definīciju, citus
nosaukumus (citās farmakopejās minētos sinonīmus) un
laboratorijas kodu;
3) attiecībā uz augu izcelsmes
preparāta nomenklatūru norāda auga binominālo zinātnisko
nosaukumu (ģints, suga, šķirne un autors) un ķīmisko tipu
(attiecīgā gadījumā), augu daļas, augu izcelsmes preparāta
definīciju, augu izcelsmes vielas attiecību pret augu izcelsmes
preparātu, ekstrakcijas šķīdinātāju(-us), citus nosaukumus (citās
farmakopejās minētos sinonīmus) un laboratorijas kodu;
4) lai dokumentētu augu izcelsmes
vielas(-u) un augu izcelsmes preparāta(-u) struktūru, norāda
fizikālo formu, to komponentu aprakstu, kuru terapeitiskā
iedarbība ir zināma, vai marķieru (molekulformula, relatīvā
molekulmasa, struktūrformula, arī relatīvā un absolūtā
stereoķīmija, molekulformula un relatīvā molekulmasa) aprakstu,
kā arī cita(-u) komponenta(-u) aprakstu;
5) lai dokumentētu sadaļu par augu
izcelsmes vielas ražotāju, sniedz visu piegādātāju (arī
līgumslēdzēju), kā arī visu paredzēto augu izcelsmes vielas
ražošanā, savākšanā un testēšanā iesaistīto ražotņu vai objektu
nosaukumu, adresi un atbildību;
6) lai dokumentētu sadaļu par augu
izcelsmes preparāta ražotāju, sniedz visu ražotāju (arī
līgumslēdzēju), kā arī visu paredzēto augu izcelsmes preparāta
ražošanā un testēšanā iesaistīto ražotņu vai objektu nosaukumu,
adresi un atbildību;
7) attiecībā uz augu izcelsmes
vielas ražošanas procesa un procesa vadības aprakstu sniedz
informāciju, lai pienācīgi aprakstītu augu ražošanu un augu
savākšanu, arī ārstniecības augu ģeogrāfisko izcelsmi un
audzēšanas, novākšanas, kaltēšanas un glabāšanas apstākļus;
8) attiecībā uz augu izcelsmes
preparāta ražošanas procesa un procesa kontroles aprakstu sniedz
informāciju, kas pienācīgi apraksta augu izcelsmes preparāta
ražošanas procesu (arī apstrādes, šķīdinātāju un reaģentu,
attīrīšanas posmu un standartizācijas aprakstus);
9) attiecībā uz ražošanas procesa
uzlabošanu sniedz konspektīvu pārskatu par augu izcelsmes
vielas(-u) un augu izcelsmes preparāta(-u) apstrādi, ņemot vērā
lietošanu un paredzēto lietošanas veidu. Ja nepieciešams,
salīdzina augu izcelsmes vielas(-u) un augu izcelsmes
preparāta(-u) fitoķīmisko sastāvu (kas izmantots
bibliogrāfiskajos datos) ar augu izcelsmes vielu(-ām) un augu
izcelsmes preparātu(-iem), kas kā aktīvā(-ās) viela(-as) ietilpst
augu izcelsmes zālēs, par ko iesniegts reģistrācijas
pieprasījums;
10) attiecībā uz augu izcelsmes
vielas struktūras un citu raksturlielumu izskaidrojumu, ja
nepieciešams, sniedz informāciju par botānisko, makroskopisko,
mikroskopisko, fitoķīmisko raksturojumu un bioloģisko
aktivitāti;
11) attiecībā uz augu izcelsmes
preparāta struktūras un citu raksturlielumu izskaidrojumu, ja
nepieciešams, sniedz informāciju par fitoķīmisko un
fizikālķīmisko raksturojumu un bioloģisko aktivitāti;
12) ja nepieciešams, sniedz augu
izcelsmes vielas(-u) un augu izcelsmes preparāta(-u)
specifikācijas;
13) ja nepieciešams, norāda augu
izcelsmes vielas(-u) un augu izcelsmes preparāta(-u) analītiskās
procedūras;
14) attiecībā uz analītisko
procedūru validāciju sniedz informāciju par analītisko
validāciju, iekļaujot augu izcelsmes vielas(-u) un augu izcelsmes
preparāta(-u) testēšanā izmantoto analītisko procedūru
eksperimentālos datus;
15) attiecībā uz sēriju analīzi
sniedz augu izcelsmes vielas(-u) un augu izcelsmes preparāta(-u)
sēriju un sēriju analīžu rezultātu aprakstu (arī par farmakopejā
iekļautajām vielām);
16) ja nepieciešams, sniedz augu
izcelsmes vielas(-u) un augu izcelsmes preparāta(-u)
specifikāciju pamatojumu;
17) ja nepieciešams, sniedz
informāciju par augu izcelsmes vielas(-u) un augu izcelsmes
preparāta(-u) testēšanā izmantotajiem references standartiem vai
materiāliem;
18) ja augu izcelsmes viela vai
augu izcelsmes preparāts ir iekļauts monogrāfijā, reģistrācijas
pieprasījuma iesniedzējs var pieprasīt Eiropas Zāļu kvalitātes
direktorāta izdotu atbilstības sertifikātu.
(2) Attiecībā uz zāļu formas
izstrādi sniedz konspektīvu pārskatu par augu izcelsmes zāļu
izstrādi, ņemot vērā paredzēto lietošanas veidu un lietošanu. Ja
nepieciešams, salīdzina produktu fitoķīmisko sastāvu (kas
izmantots bibliogrāfiskajos datos) ar augu izcelsmes zālēm, par
ko iesniegts reģistrācijas pieprasījums.
61. Zāles reti sastopamu slimību
ārstēšanai:
(1) Zālēm reti sastopamu slimību
ārstēšanai piemēro Eiropas Parlamenta un Padomes 1999.gada
16.decembra Regulas (EK) Nr.141/2000 nosacījumus par
medikamentiem reti sastopamu slimību ārstēšanai vai šī pielikuma
53.punkta vispārīgos noteikumus. Reģistrācijas pieprasījuma
iesniedzējs neklīniskās un klīniskās izpētes kopsavilkumos pamato
iemeslus, kādēļ nav iespējams sniegt pilnīgu informāciju, un
pamato attiecīgo zāļu reti sastopamu slimību ārstēšanai ieguvumu
un riska attiecību.
(2) Ja tādas reģistrācijas
pieprasījuma iesniedzējs neņem vērā šo noteikumu 19.2.apakšpunkta
un šī pielikuma 48.punkta nosacījumus saskaņā ar Farmācijas
likuma 10.panta 7.punktā nosacījumiem zālēm, ko piegādā, izpildot
bona fide pasūtījumu (pasūtījuma izdarīšana nav
veicināta), un kas izgatavotas saskaņā ar pilnvarota veselības
aprūpes speciālista specifikācijām un ir paredzētas lietošanai
konkrētiem pacientiem, viņam uzņemoties tiešu personisku
atbildību, sistemātisko un dokumentēto attiecīgās vielas
lietošanu var attiecināt uz minētās vielas lietošanu.
D sadaļa
Uzlabotas terapeitiskās iedarbības zāles
62. Šajā sadaļā noteiktas prasības
uzlabotas terapeitiskās iedarbības zālēm:
(1) Gēnu (cilvēka un ksenogēnu)
terapijas zāles:
1) gēnu terapijas zāļu
dažādība;
2) īpašas prasības attiecībā uz
3.moduli.
(2) Somatisko šūnu (cilvēka un
ksenogēnu) terapijas zāles.
(3) Īpašas prasības gēnu terapijas
un šūnu terapijas (cilvēka un ksenogēnu) zālēm saistībā ar 4. un
5.moduli:
1) 4.modulis;
2) 5.modulis:
a) farmakoloģijas un efektivitātes
pētījumi ar cilvēkiem;
b) drošība.
(4) Īpašas prasības
ksenotransplantācijas zālēm.
63. Uzlabotas terapeitiskās
iedarbības zāles iegūst ražošanas procesos, kur par aktīvajām
vielām vai aktīvo vielu daļu izmanto galvenokārt dažādas gēnu
pārneses ceļā radītas biomolekulas, kā arī bioloģiski uzlabotas
terapeitiski pārveidotas šūnas. Šādām zālēm reģistrācijas
pieprasījuma dokumentācijas noformējums atbilst šī pielikuma A
sadaļas prasībām. Piemēro 1., 2., 3., 4. un 5.moduli. Attiecībā
uz ģenētiski modificētu organismu apzinātu izplatīšanu vidē
uzmanību pievērš ģenētiski modificēto organismu noturībai
recipientā un arī ģenētiski modificēto organismu replikācijai, kā
arī pārveidojumiem, tos izplatot vidē. Informāciju par vides
risku atspoguļo 1.moduļa pielikumā.
64. Gēnu terapijas zāles (cilvēku
un ksenogēnas) šīs sadaļas izpratnē ir produkts, kas iegūts no
tāda ražošanas procesu kopuma, kuru mērķis ir pārnest
profilaktisku, diagnostisku vai terapeitisku gēnu (nukleīnskābes
daļu) uz cilvēka vai uz dzīvnieka šūnām in vivo vai ex
vivo, kā arī tā izpausme in vivo. Gēnu pārnese ir
ekspresijas sistēma, kas ietilpst pārneses sistēmā, ko pazīst kā
vektoru, kas var būt vai nebūt vīrusu izcelsmes. Vektors var būt
iekļauts arī cilvēka vai dzīvnieka šūnā.
65. Gēnu terapijas zāles ir:
(1) Zāles, kuru pamatā ir alogēnas
vai ksenogēnas šūnas. Vektors pirms pārneses uz saimniekšūnām ir
gatavs un tiek glabāts. Šūnas ir iegūtas iepriekš un var tikt
apstrādātas kā šūnu banka (bankas kolekcijas vai banka, kas
izveidota no cilmes šūnām) ar ierobežotu dzīvotspēju. Šūnas, kas
ģenētiski modificētas ar vektoru, ir aktīvā viela. Gatavo zāļu
iegūšanai var veikt papildu darbības. Pēc būtības tādas zāles ir
paredzēts lietot noteiktam skaitam pacientu.
(2) Zāles, kurās izmantotas
autologas cilvēka šūnas. Aktīvā viela ir gatavs vektoru kopums,
kas pirms pārneses uz autologām šūnām tiek glabāts. Lai iegūtu
gatavās zāles, var veikt papildu darbības. Šīs zāles gatavo no
konkrēta pacienta iegūtām šūnām. Vēlāk šūnas tiek ģenētiski
modificētas, izmantojot gatavu vektoru, kas satur attiecīgo gēnu,
kurš iepriekš sagatavots un veido aktīvo vielu. Preparātu injicē,
un tas ir paredzēts vienam pacientam. Visu ražošanas procesu no
šūnu savākšanas līdz injicēšanai pacientam uzskata par vienu
intervenci.
(3) Gatava vektoru lietošana ar
iespraustu (profilaktisku, diagnostisku vai terapeitisku)
ģenētisko materiālu. Aktīvā viela ir gatavu vektora kopums. Lai
iegūtu gatavās zāles, var veikt papildu darbības. Šāda veida
zāles ir paredzētas lietošanai vairākiem pacientiem. Ģenētiskā
materiāla pārnesi var veikt, gatavo vektoru tieši injicējot
recipientiem.
66. Īpašas prasības attiecībā uz
3.moduli gēnu terapijas zālēm:
(1) Gēnu terapijas zāles ir
šādas:
1) kailā nukleīnskābe;
2) kompleksā nukleīnskābe vai
vektori, kas nav vīrusu vektori;
3) vīrusu vektori;
4) ģenētiski modificētas
šūnas.
(2) Attiecībā uz citām zālēm
izšķir trīs galvenos ražošanas procesa faktorus:
1) izejvielas — materiāli, no
kā ražo aktīvo vielu (piemēram, attiecīgais gēns, ekspresijas
plazmīdas, šūnu un vīrusu bankas vai vektori, kas nav vīrusu
vektori);
2) aktīvā viela —
rekombinantais vektors, vīruss, kailās vai kompleksās plazmīdas,
vīrusus ražojošās šūnas, in vitro ģenētiski modificētas
šūnas;
3) gatavās zāles — aktīvā
viela galīgajā tiešajā iepakojumā izmantošanai medicīnā. Atkarībā
no gēnu terapijas zāļu veida, lietošanas veida un nosacījumiem
var būt nepieciešama pacienta šūnu ārstēšana ex vivo
(skatīt 65.panta otro daļu).
(3) Īpašu uzmanību pievērš šādiem
jautājumiem:
1) sniedz informāciju par gēnu
terapijas zāļu attiecīgajiem rādītājiem, tajā skaitā par to
izpausmi mērķa šūnu populācijā. Sniedz informāciju par kodētu
gēnu secības avotu, uzbūvi, raksturojumu un verifikāciju, tajā
skaitā par tās viengabalainību un stabilitāti. Papildus
terapeitiskajam gēnam norāda arī citu gēnu pilnīgu secību,
regulētājelementus un vektora pamatuzbūvi;
2) sniedz informāciju par tā
vektora raksturojumu, kas izmantots gēna pārnesei un uzņemšanai.
Iekļauj tā fizikālķīmisko, bioloģisko un imunoloģisko
raksturojumu. Par zālēm, kam gēnu pārnesei izmanto mikroorganismu
(piemēram, baktērijas vai vīrusus (bioloģiskā gēnu pārnese)),
sniedz datus par vecāku celma patoģenēzi un par tā tropismu uz
īpašiem audiem un šūnu tipiem, kā arī mijiedarbības šūnu atkarību
no cikla. Par zālēm, kam gēnu pārnesei neizmanto bioloģiskus
līdzekļus, norāda atsevišķu komponentu un to kombināciju
fizikālķīmiskās īpašības;
3) šūnu banku vai sējmateriāla
sērijas izveidošanas principus un raksturojumu pēc vajadzības
attiecina uz gēnu terapijas zālēm;
4) norāda rekombinantā vektora
saimniekšūnu avotu. Dokumentē cilvēka raksturojumu (piemēram,
vecumu, dzimumu, mikrobioloģisko un vīrusu analīžu rezultātus,
izslēgšanas kritērijus un izcelsmes valsti). Par dzīvnieku
izcelsmes šūnām norāda detalizētu informāciju šādos
jautājumos:
a) dzīvnieku izcelsmes
noteikšana;
b) lopkopība un aprūpe;
c) transgēnie dzīvnieki
(izveidošanas metodes, transgēno šūnu raksturojums, iesprausto
gēnu īpašības);
d) pasākumi izcelsmes (donoru)
dzīvnieku infekciju novēršanai un uzraudzībai;
e) testēšana attiecībā uz
infekcijas izraisītājiem;
f) aprīkojums;
g) izejvielu un neapstrādāto
materiālu kontrole;
5) dokumentē šūnu savākšanas
metožu aprakstu, atrašanās vietu, audu veidu, apstrādes procesu,
transportēšanu, glabāšanu un izsekojamību, kā arī savākšanas
procesa laikā veiktās pārbaudes;
6) vīrusu drošības novērtējums, kā
arī produktu izsekojamība no donora līdz gatavajām zālēm ir
būtiska iesniedzamās dokumentācijas daļa (piemēram, jāizslēdz
replicēties spējīgu vīrusu klātbūtne replicēties nespējīgu vīrusu
vektoru bankās).
67. Somatisko šūnu terapijas zāles
(cilvēku un ksenogēnu) ir autologas (no paša pacienta), alogēnas
(no cita cilvēka) vai ksenogēnas (no dzīvniekiem) somatiskas
dzīvas šūnas, ko izmanto cilvēkiem un kuru bioloģiskais
raksturojums ir būtiski mainīts, ar tām manipulējot, lai gūtu
terapeitisku, diagnostisku vai profilaktisku iedarbību ar
metaboliskiem, farmakoloģiskiem vai imunoloģiskiem līdzekļiem.
Šajā manipulācijā iekļauta autologo šūnu populāciju paplašināšana
vai aktivizēšana ex vivo (piemēram, adoptīvā
imūnterapija), ar medicīnas ierīcēm saistītu alogēno vai
ksenogēno šūnu lietošana ex vivo vai in vivo
(piemēram, mikrokapsulas, kompleksās matricas, kas bioloģiski
noārdās vai nenoārdās). Īpašas prasības šūnu terapijas zālēm
attiecībā uz 3.moduli:
67.1. somatisko šūnu terapijas
zāles ir šādas:
(1) Šūnas, ar ko manipulē, lai
kvalitatīvi vai kvantitatīvi pārveidotu to imunoloģiskās,
metaboliskās vai citas funkcionālās īpašības.
(2) Šūnas, ko šķiro, atlasa un ar
ko manipulē, lai vēlāk ražošanas procesā iegūtu gatavās
zāles.
(3) Šūnas, ar ko manipulē un ko
kombinē ar bezšūnu komponentiem (piemēram, bioloģiskās vai
inertās matricas vai medicīnas ierīces), un kas parāda vielas
paredzēto darbību gatavajās zālēs.
(4) Autologo šūnu atvasinājumi,
kas īpašu kultūru apstākļos izpaužas in vitro.
(5) Šūnas, kas ir ģenētiski
modificētas vai ar ko izdarītas citādas manipulācijas, lai tām
izpaustos iepriekš neizpaudušās homoloģiskas vai nehomoloģiskas
funkcionālās īpašības.
67.2. visu ražošanas procesu no
šūnu savākšanas (autologa situācija) līdz pat injicēšanai
pacientam uzskata par vienu intervenci;
67.3. citām zālēm nosaka šādus
trīs ražošanas procesa elementus:
(1) Izejvielas — vielas, no
kā ražo aktīvo vielu (orgāni, audi, ķermeņa šķidrumi vai
šūnas).
(2) Aktīvā viela — šūnas, ar
ko izdarītas manipulācijas, šūnu lizāti, augoša šūnu kultūra un
šūnas, ko izmanto kopā ar inertām matricām vai medicīnas
ierīcēm.
(3) Gatavās zāles — aktīvā
viela tās galīgajā tiešajā iepakojumā izmantošanai medicīnā:
1) vispārīga informācija par
aktīvo(-ajām) vielu(-ām). Šūnu terapijas zāļu aktīvās vielas
sastāv no šūnām, kas pēc pārstrādāšanās in vitro uzrāda
profilaktiskas, diagnostiskas vai terapeitiskas īpašības, kas
atšķiras no sākotnējām fizioloģiskajām vai bioloģiskajām
īpašībām. Šajā sadaļā apraksta attiecīgo šūnu un kultūru veidu.
Dokumentē audus, orgānus vai bioloģiskos šķidrumus, no kā šūnas
iegūtas, kā arī donoru materiālu autologās, alogēnās vai
ksenogēnās īpašības un to ģeogrāfisko izcelsmi. Detalizēti
apraksta šūnu savākšanu, paraugu atlasi un glabāšanu pirms
turpmākas apstrādes. Attiecībā uz alogēnajām šūnām īpašu uzmanību
pievērš pirmajiem procesa posmiem, kuros ietilpst donoru atlase.
Norāda veikto manipulāciju veidu, par aktīvo vielu izmantoto šūnu
fizioloģisko funkciju;
2) informācija par aktīvās(-o)
vielas(-u) izejmateriāliem (cilvēka un dzīvnieku somatiskās
(ksenogēnās) šūnas).
68. Šī pielikuma 67.punktā minētās
cilvēka somatiskās šūnu terapijas zāles gatavo no noteikta skaita
dzīvotspējīgu šūnu (šūnu fonds), ko iegūst ražošanas procesā, kas
sākas no cilvēka iegūtu orgānu vai audu līmenī, vai noteiktas
šūnu bankas sistēmas līmenī, kur šūnu fonda pamatā ir
nepārtrauktas šūnu līnijas. Šajā nodaļā aktīvā viela ir cilvēka
šūnu kultūras fonds, bet gatavās zāles ir cilvēka šūnu kultūras
fonds, kas sagatavots paredzētajai lietošanai medicīnā. Pilnībā
dokumentē izejvielas un visus ražošanas procesa posmus, kā arī
vīrusu drošības aspektus:
(1) Cilvēka izcelsmes orgāni,
audi, ķermeņa šķidrumi un šūnas. Dokumentē cilvēka avota
raksturojumu, piemēram, vecumu, dzimumu, mikrobioloģisko
stāvokli, izslēgšanas kritērijus un izcelsmes valsti. Dokumentē
paraugu atlases aprakstu, vietu, veidu, apstrādes procesu,
fondus, transportēšanu, glabāšanu un izsekojamību, kā arī veiktās
atlasīto paraugu pārbaudes.
(2) Šūnu banku sistēmas.
Attiecīgās šajā pielikumā aprakstītās prasības attiecina uz šūnu
banku sistēmas sagatavošanu un kvalitātes kontroli. Tas var
attiekties uz alogēnajām vai ksenogēnajām šūnām.
(3) Palīgmateriāli vai medicīnas
palīgierīces. Sniedz informāciju par visu neapstrādāto materiālu
izmantošanu (piemēram, citokīniem, augšanas faktoriem, barotnēm)
vai par iespējamiem palīgproduktiem un medicīnas ierīcēm
(piemēram, šūnu šķirošanas ierīcēm, bioloģiski saderīgiem
polimēriem, matricām, šķiedrām, pērlītēm un to bioloģisko
saderību, funkcionalitāti, kā arī par infekcijas izraisītāju
risku).
69. Par šī pielikuma 67.punktā
minētām dzīvnieku somatiskām šūnām (ksenogēnās) sniedz šādu
detalizētu informāciju:
(1) Dzīvnieku izcelsmes
noteikšana.
(2) Lopkopība un aprūpe.
(3) Ģenētiski modificēti dzīvnieki
(izveidošanas metodes, transgēno šūnu raksturojums, iesprausto
vai izņemto gēnu īpašības).
(4) Pasākumi izcelsmes, kā arī
donoru dzīvnieku infekciju novēršanai un pārraudzībai.
(5) Testēšana attiecībā uz
infekcijas izraisītājiem, tajā skaitā par vertikāli pārnestajiem
mikroorganismiem (arī endogēnajiem retrovīrusiem).
(6) Aprīkojums.
(7) Šūnu banku sistēmas.
(8) Izejvielu un neapstrādāto
materiālu kontrole.
70. Par šo noteikumu 68.punktā
minētajām zālēm sniedz šādu informāciju:
(1) Aktīvās(-o) vielas(-u) un
gatavā produkta ražošanas process. Dokumentē dažādos ražošanas
procesa posmus (piemēram, orgānu (audu) disociāciju, attiecīgo
šūnu populācijas atlasi, in vitro šūnu kultūru, šūnu
transformāciju ar fizikālķīmiskiem aģentiem vai gēnu
pārnesi).
(2) Aktīvās(-o) vielas(-u)
raksturojumā sniedz informāciju par attiecīgo šūnu populācijas
identitāti (izcelsmes šūnas, saistītā citoģenētika, morfoloģiskā
analīze), tīrību (nejaušas mikrobu vielas un šūnu piemaisījumi),
potenci (noteikta bioloģiskā aktivitāte) un atbilstību
(karioloģijas un tumoroģenēzes testi) paredzētajai lietošanai
medicīnā.
(3) Gatavo zāļu farmaceitiskā
izstrāde. Papildus izmantotajai īpašajai lietošanas metodei
(intravenozā infūzija, lokālā injekcija, transplantācijas
ķirurģija) sniedz informāciju arī par iespējamo medicīnas
palīgierīču izmantošanu (bioloģiski saderīgi polimēri, matricas,
šķiedras, pērlītes) un to bioloģisko saderību un izturību.
(4) Izsekojamība. Sniedz
detalizēti izstrādātu shēmu, kas nodrošina produktu izsekojamību
no donora līdz gatavajām zālēm.
71. Īpašas prasības gēnu terapijas
un šūnu terapijas (cilvēku un ksenogēnu) zālēm saistībā ar:
71.1. 4.moduli:
(1) Ir zināms, ka 4.modulī
noteiktās prasības par zāļu neklīnisko pārbaudi ne vienmēr ir
piemērotas attiecībā uz gēnu un somatisko šūnu terapijas zālēm to
unikālās un daudzveidīgās struktūras un bioloģisko īpašību dēļ,
tai skaitā sugu izteiktā specifiskuma, subjektu specifiskuma,
imunoloģiso barjeru un atšķirīgo plejotropisko atbilžu dēļ.
(2) Motīvus, kas pamato neklīnisko
izstrādi, un kritērijus, kas izmantoti attiecīgu sugu un modeļu
izvēlei, apraksta 2.modulī.
(3) Var noteikt vai izstrādāt
jaunus izpētes modeļus dzīvniekiem, lai zāļu pētījumos in
vivo par darbību cilvēka organismā palīdzētu ekstrapolēt
īpašus datus funkcionāliem mērķa raksturlielumiem un
toksicitātei. Sniedz zinātnisko pamatojumu attiecībā uz šo
slimības modeļu izmantošanu dzīvniekiem, lai pamatotu koncepcijas
par efektivitāti drošību un ticamību.
71.2. 5.moduli:
(1) Uzlabotas terapeitiskās
iedarbības zāļu efektivitāti pierāda, kā noteikts 5.modulī. Dažām
zālēm un dažām terapeitiskajām indikācijām var būt neiespējami
veikt parasto klīnisko izpēti. Atkāpes no spēkā esošajām
pamatnostādnēm pamato 2.modulī. Zāļu klīniskajai izstrādei ir
dažas īpašas pazīmes, jo aktīvajām vielām ir komplicētas un
labilas īpašības. Nepieciešami papildu apsvērumi par šūnu
dzīvotspēju, proliferāciju, migrāciju un diferenciāciju
(somatisko šūnu terapija), par īpašajiem klīniskajiem apstākļiem,
kādos zāles lieto, vai par gēnu ekspresijas darbības īpašo veidu
(somatisko gēnu terapija). Zāļu reģistrācijas pieprasījumā norāda
ar minētajiem produktiem saistītos riskus, ko var radīt iespējami
infekcijas izraisītāju piemaisījumi. Īpašu uzmanību pievērš
izstrādes agrīnajiem posmiem, tajā skaitā donoru izvēlei šūnu
terapijas zālēm, visai terapeitiskajai intervencei kopumā, zāļu
pareizai apstrādei un lietošanai. Reģistrācijas pieprasījums
5.modulī iekļauj attiecīgos datus par pasākumiem, ko veic, lai
uzraudzītu un kontrolētu dzīvo šūnu funkciju un attīstību
recipientā, lai novērstu infekcijas izraisītāju pārnešanu uz
recipientu un lai līdz minimumam samazinātu iespējamo risku
sabiedrības veselībai:
1) farmakoloģijas un efektivitātes
pētījumi ar cilvēkiem:
a) par farmakoloģijas pētījumiem
ar cilvēkiem sniedz informāciju par paredzēto darbības veidu un
paredzēto efektivitāti saskaņā ar pamatotiem mērķa
raksturlielumiem, biosadalījumu, atbilstošu devu, grafiku un
lietošanas paņēmieniem vai lietošanas veidu, kāds vēlams
efektivitātes pētījumos;
b) farmakokinētikas pētījumi var
būt nebūtiski attiecībā uz dažām uzlabotas terapeitiskās
iedarbības zālēm, ja pētījumi ar veseliem brīvprātīgajiem nav
iespējami un noteikt devu un kinētiku klīniskajos pētījumos ir
grūti. Pēta produkta izplatīšanos un darbību in vivo, tajā
skaitā šūnu proliferāciju un darbību ilgā laikā, kā arī gēnu
preparāta apmēru, izplatīšanos un vēlamās gēnu ekspresijas
ilgumu. Veic un vajadzības gadījuma izstrādā atbilstošus testus,
lai izsekotu šūnu preparātu vai šūnu, kas cilvēka ķermenī izpauž
vēlamo gēnu, kā arī lai uzraudzītu lietoto vai transficēto šūnu
darbību;
c) uzlabotas terapeitiskās
iedarbības zāļu efektivitātes un drošības novērtējumā iekļauj
terapeitiskās procedūras aprakstu un novērtējumu kopumā īpašiem
lietošanas veidiem (piemēram, šūnu transfekcija ex vivo,
in vitro manipulācijas vai intervences metodes), kā arī
iespējamo saistīto režīmu pārbaudes (tajā skaitā imunitāti
vājinoša, pretvīrusu, citotoksiskā apstrāde);
d) procedūru testē klīniskajā
izpētē un apraksta zāļu aprakstā;
2) drošība:
a) izskata drošības jautājumus,
kas saistīti ar imūnreakciju uz zālēm vai uz izpaustajiem
proteīniem, imūno atgrūšanu, imūndepresiju un imūnizolācijas
mehānismu iedalījumu;
b) dažas uzlabotas gēnu terapijas
zāles un somatisko šūnu terapijas zāles (piemēram, ksenogēno šūnu
terapijas un dažas gēnu pārneses zāles) var saturēt daļiņas, kas
spēj replicēties, un/vai infekcijas izraisītājus. Novēro, vai
pacientam neattīstās iespējamas infekcijas, kā arī patoloģiskas
komplikācijas pirmsatļaujas, kā arī pēcatļaujas posmā. Ja
nepieciešams, minēto uzraudzību attiecina arī uz cilvēkiem, kam
ir saskare ar pacientu, kā arī uz veselības aprūpes
darbiniekiem;
c) ja lieto dažas somatisko šūnu
terapijas zāles un dažas gēnu pārneses zāles, pilnībā nevar
izslēgt iespējami transmisīvu vielu piemaisījumu risku, bet ar
3.modulī aprakstītajiem attiecīgajiem pasākumiem risku var
mazināt;
d) ražošanas procesā iekļautos
pasākumus papildina ar testēšanas metodēm, kvalitātes kontroles
procesiem un atbilstošām uzraudzības metodēm, kas jāapraksta
5.modulī;
e) dažu uzlabotu somatisko šūnu
terapijas zāļu lietošana uz laiku vai pastāvīgi var būt atļaujama
tikai iestādēs, kurās ir dokumentēta ekspertīze un iekārtas, kas
nodrošina īpašu pacientu drošības uzraudzību. Līdzīga pieeja
iespējama noteiktām gēnu terapijas zālēm, ko saista ar tādu
infekcijas izraisītāju iespējamo risku, kas spēj replicēties;
f) attiecīgā gadījumā
reģistrācijas pieprasījumā aplūko un pievērš uzmanību arī
ilgstošas uzraudzības aspektiem par vēlīnu komplikāciju
attīstību;
g) attiecīgā gadījumā
reģistrācijas pieprasījuma iesniedzējs iesniedz detalizēti
izstrādātu riska pārvaldības plānu, iekļaujot klīniskos un
laboratorijas datus par pacientu, potenciālos epidemioloģiskos
datus un, ja tas ir būtiski, — datus no donora un recipienta
audu paraugu arhīviem. Šāda sistēma ir nepieciešama, lai
nodrošinātu zāļu izsekojamību un ātru reakciju uz šaubīgiem
nevēlamiem notikumiem.
72. Īpašas prasības
ksenotransplantācijas zālēm (ksenotransplantācija ir procedūra,
kas ietver no dzīvniekiem iegūtu dzīvu audu vai orgānu, vai ar
dzīvām dzīvnieka šūnām, audiem vai orgāniem ex vivo
saskarē bijušu cilvēka organisma šķidrumu, šūnu, audu vai orgānu
transplantāciju, implantāciju vai infūziju recipientam cilvēkam)
un to izejvielām. Sniedz detalizētu informāciju par šādiem
jautājumiem:
1) dzīvnieku izcelsmes
noteikšana;
2) lopkopība un aprūpe;
3) ģenētiski modificēti dzīvnieki
(izveidošanas metodes, transgēno šūnu raksturojums, iesprausto
vai izņemto gēnu īpašības);
4) pasākumi izcelsmes
donordzīvnieku infekciju novēršanai un uzraudzībai;
5) testēšana attiecībā uz
infekcijas izraisītājiem;
6) aprīkojums;
7) izejvielu un neapstrādāto
materiālu kontrole;
8) izsekojamība.”
“3.pielikums
Ministru kabineta
2000.gada 31.oktobra
noteikumiem Nr.381
Reģistrācijas
apliecības satura paplašināšana
Izmaiņas reģistrācijas
dokumentācijā, par ko reģistrācijas īpašnieks iesniedz iesniegumu
reģistrācijas apliecības satura paplašināšanai, ir šādas:
1. Izmaiņas, kas saistītas ar
aktīvo(-ajām) vielu(-ām):
1.1. aktīvās(-o) vielas(-u)
aizstāšana ar citu sāls vai estera kompleksu vai to atvasinājumu
(ar to pašu ārstniecisko sastāvdaļu), ja iedarbīguma, kā arī
drošības raksturlielumi būtiski neatšķiras;
1.2. aktīvās vielas aizstāšana ar
citu izomēru vai citu izomēru maisījumu, maisījuma aizstāšana ar
izolētu izomēru (piemēram, racemātu aizstāj ar vienu
enantiomēru), ja iedarbīguma, kā arī drošības raksturlielumi
būtiski neatšķiras;
1.3. bioloģiska vielas vai
biotehnoloģiska produkta aizstāšana ar vielu, kuras molekulai ir
mazliet citāda struktūra. Vektora pārveidošana, kuru izmanto
antigēna, kā arī izejmateriāla izgatavošanā (arī jauna izejas
(galveno) šūnu banka no cita avota), ja iedarbīguma, kā arī
drošības raksturlielumi būtiski neatšķiras;
1.4. jauns ligands vai savienojuma
mehānisms radiofarmaceitiskam preparātam;
1.5. izmaiņas ekstrakcijas
šķīdinātāja vai augu izcelsmes zāļu augu attiecībā, ja
iedarbīguma, kā arī drošības raksturlielumi būtiski
neatšķiras.
2. Izmaiņas stiprumā, zāļu formā
vai lietošanas veidā:
2.1. biopieejamības izmaiņas;
2.2. izmaiņas farmakokinētikā
(piemēram, atbrīvošanās ātruma pakāpē);
2.3. izmaiņas vai jauna stipruma,
kā arī iedarbības pievienošana;
2.4. izmaiņas vai jaunas zāļu
formas pievienošana;
2.5. lietošanas veida izmaiņas vai
jauna veida pievienošana. Ja zāles ievada parenterāli, norāda
ievadīšanas veidu (piemēram, intraarteriālais, intravenozais,
intramuskulārais, zemādas).”
"4.pielikums
Ministru kabineta
2000.gada 31.oktobra
noteikumiem Nr.381
Nelielas IA un IB tipa
izmaiņas

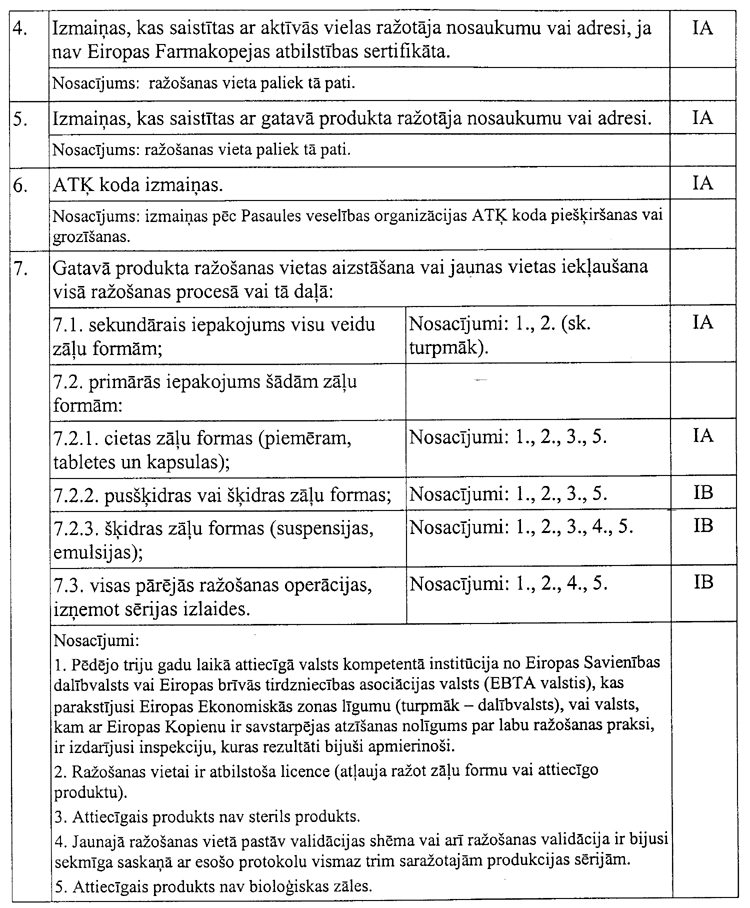
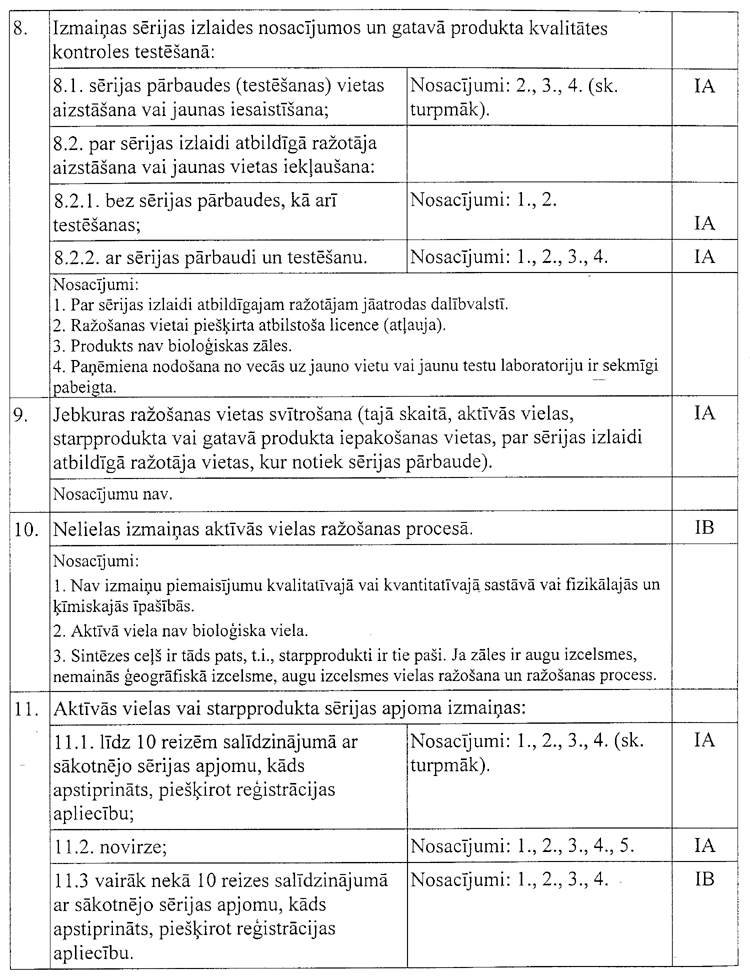
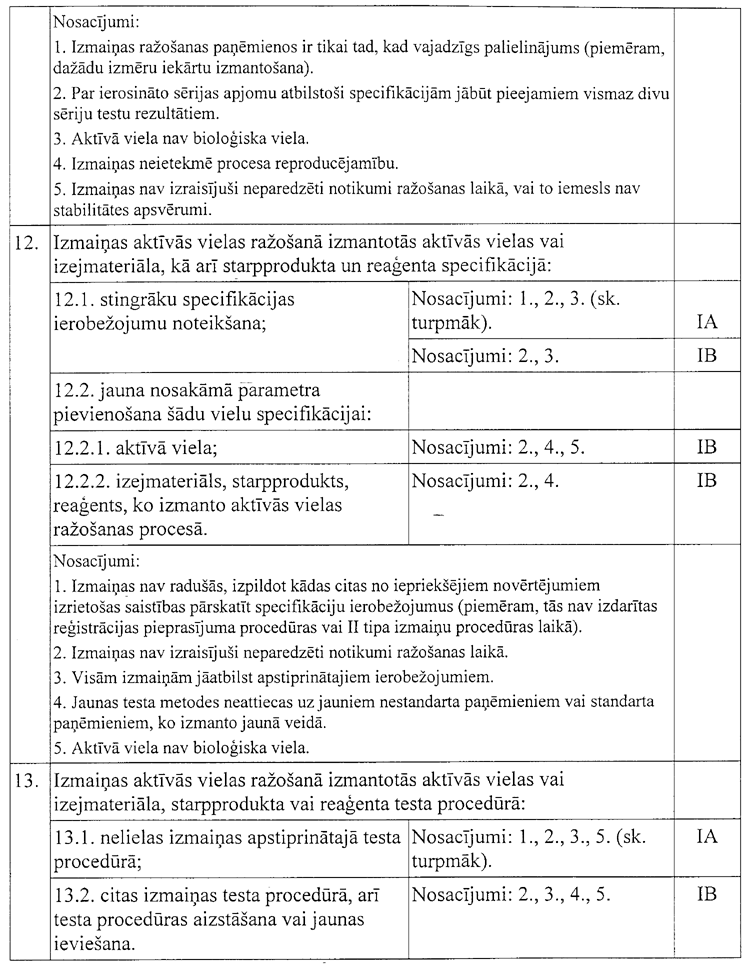
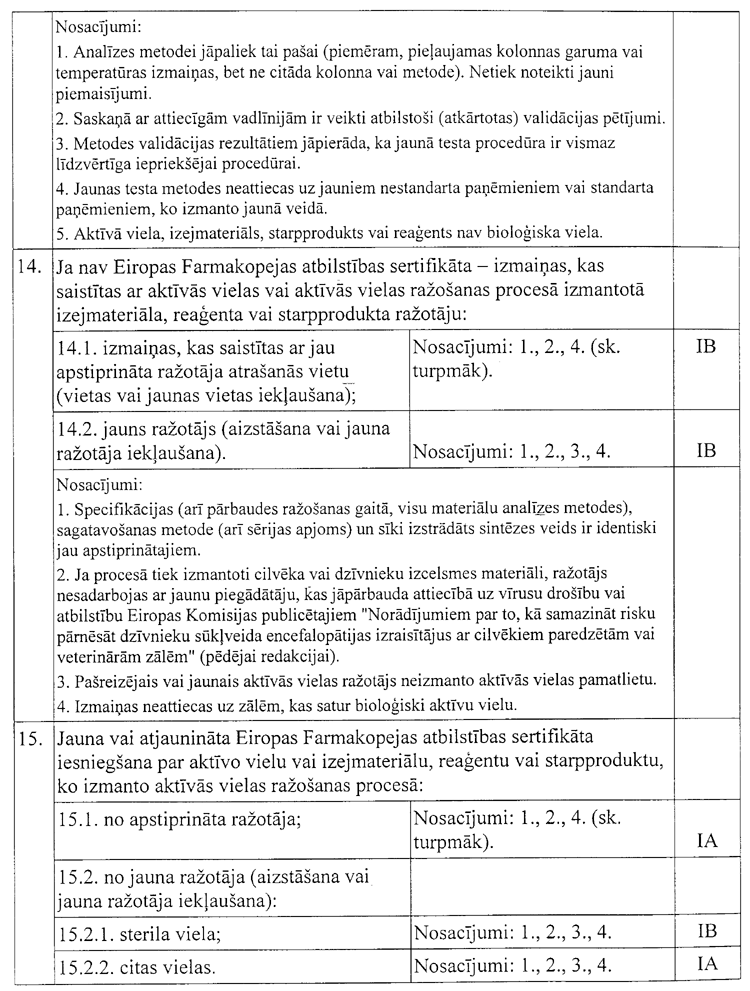
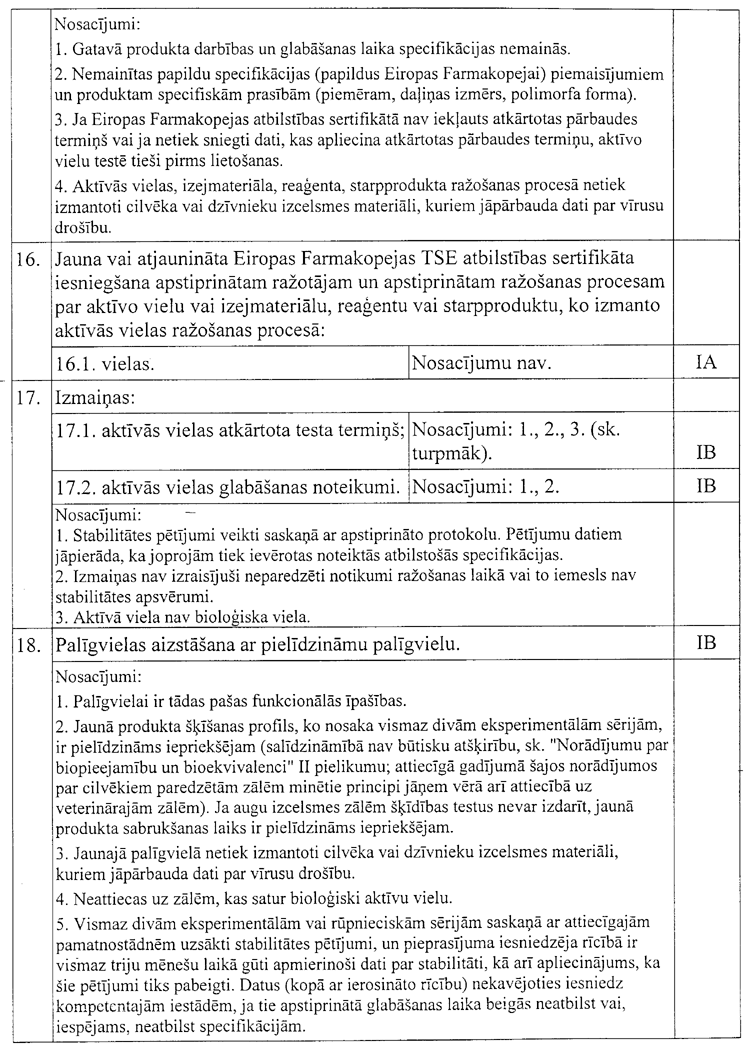
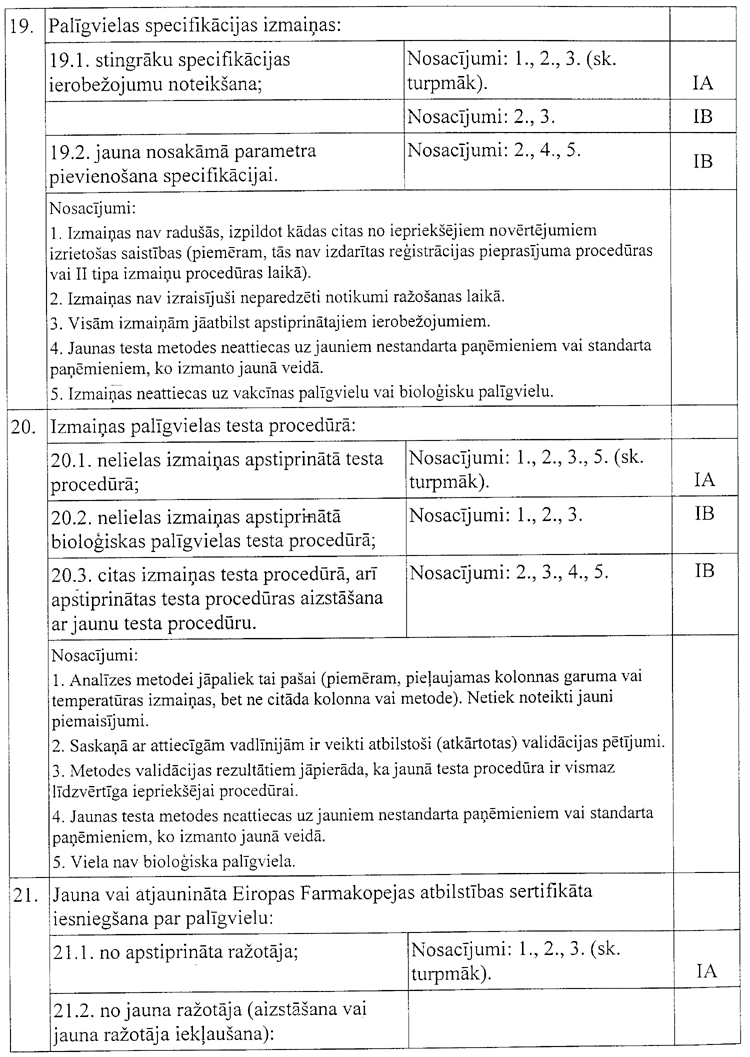
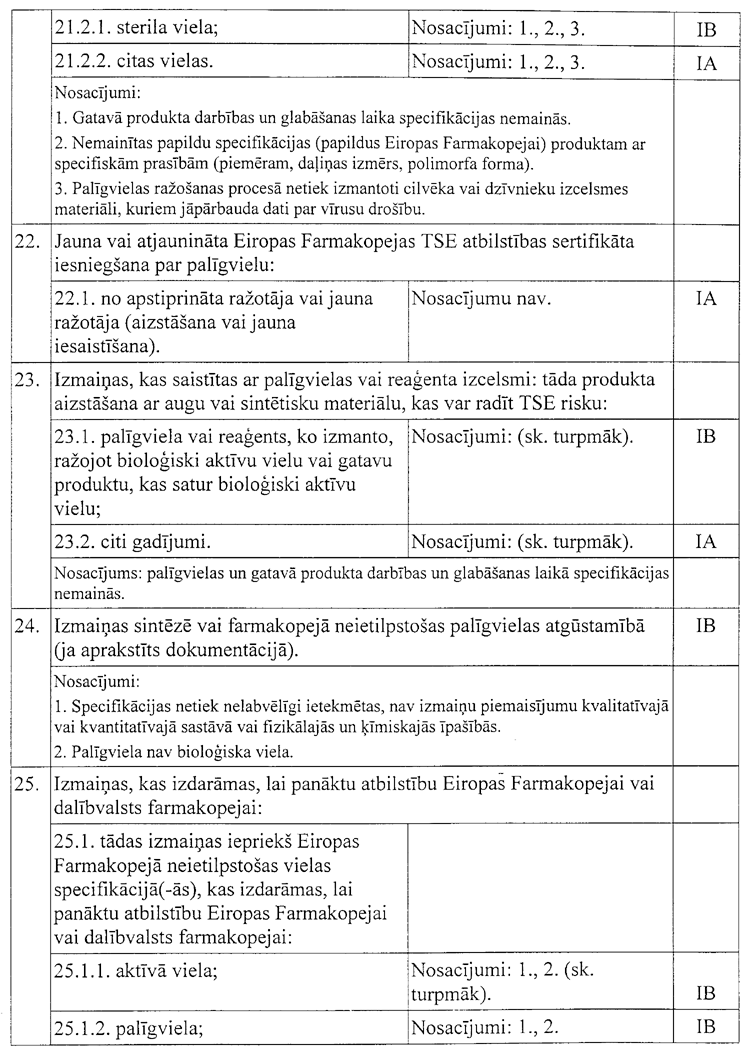
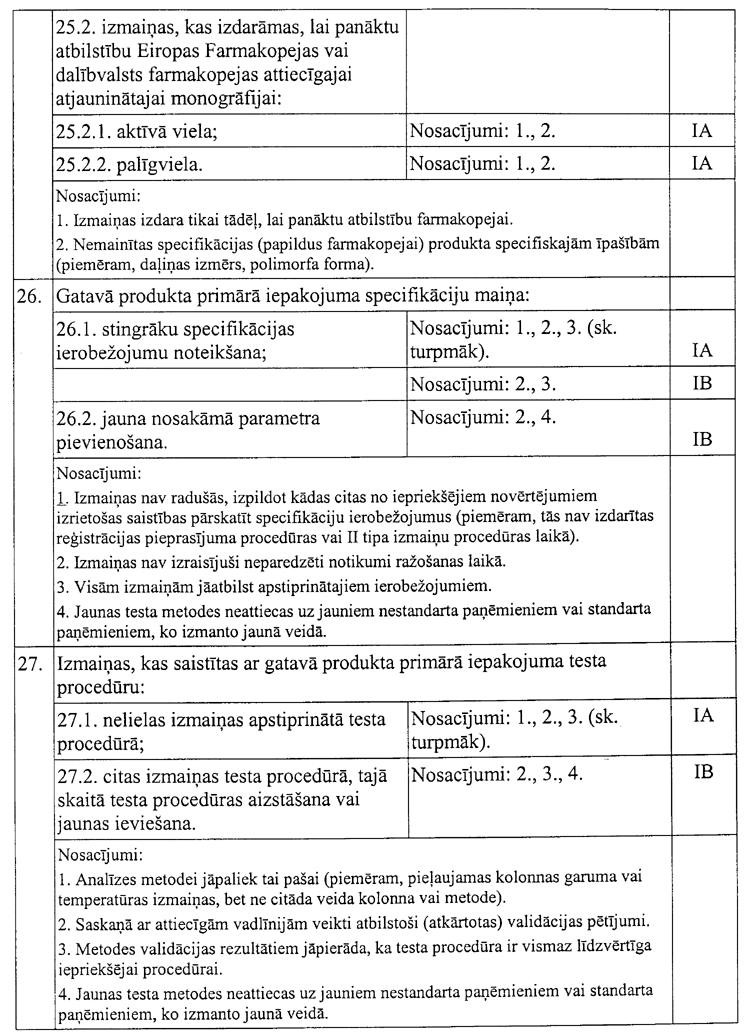
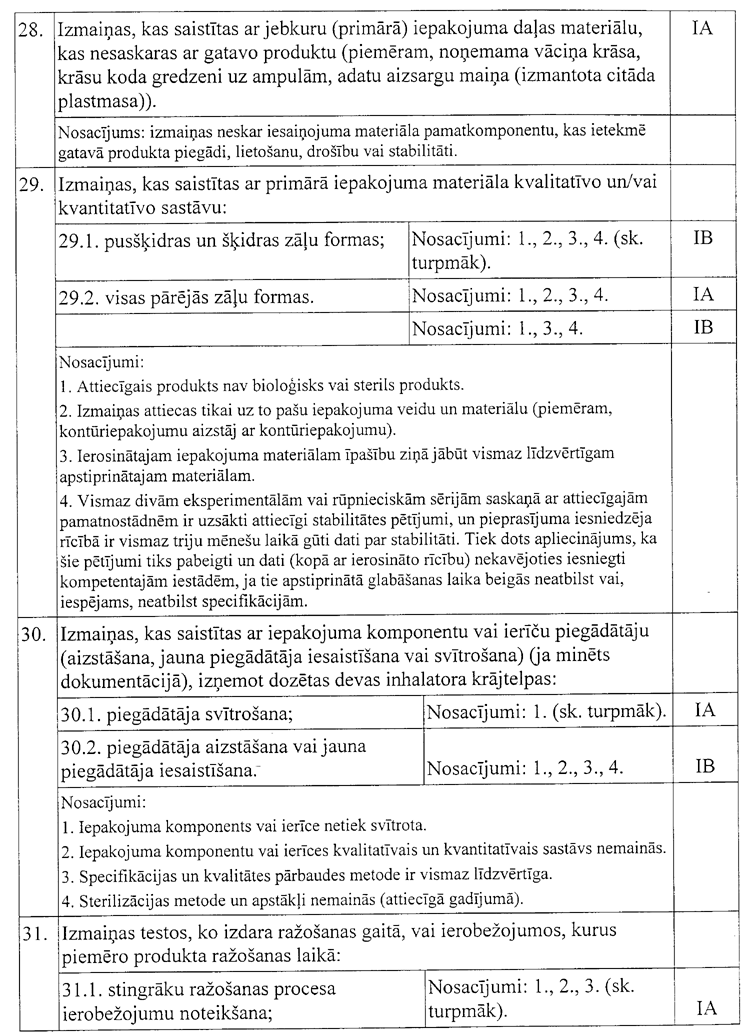

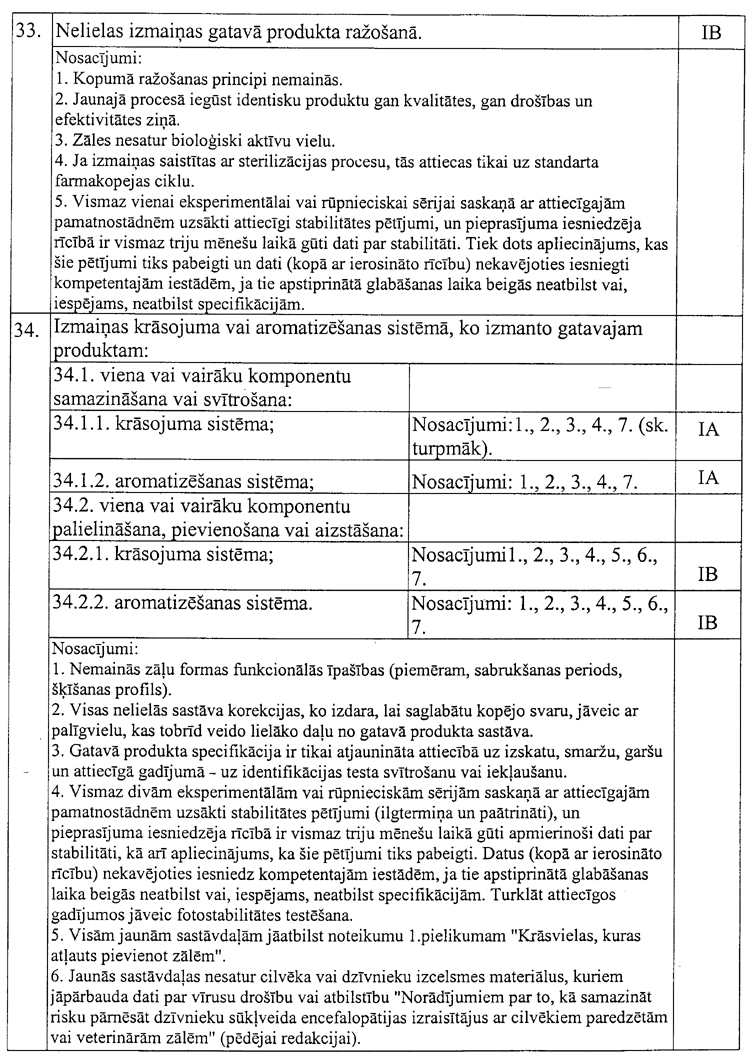
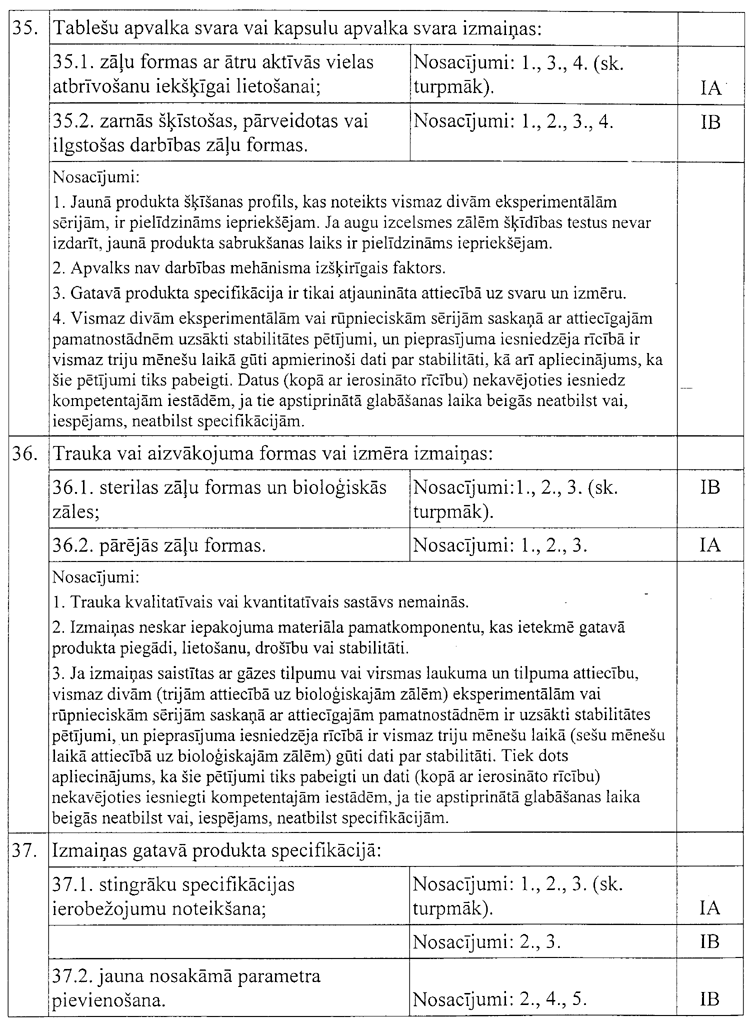
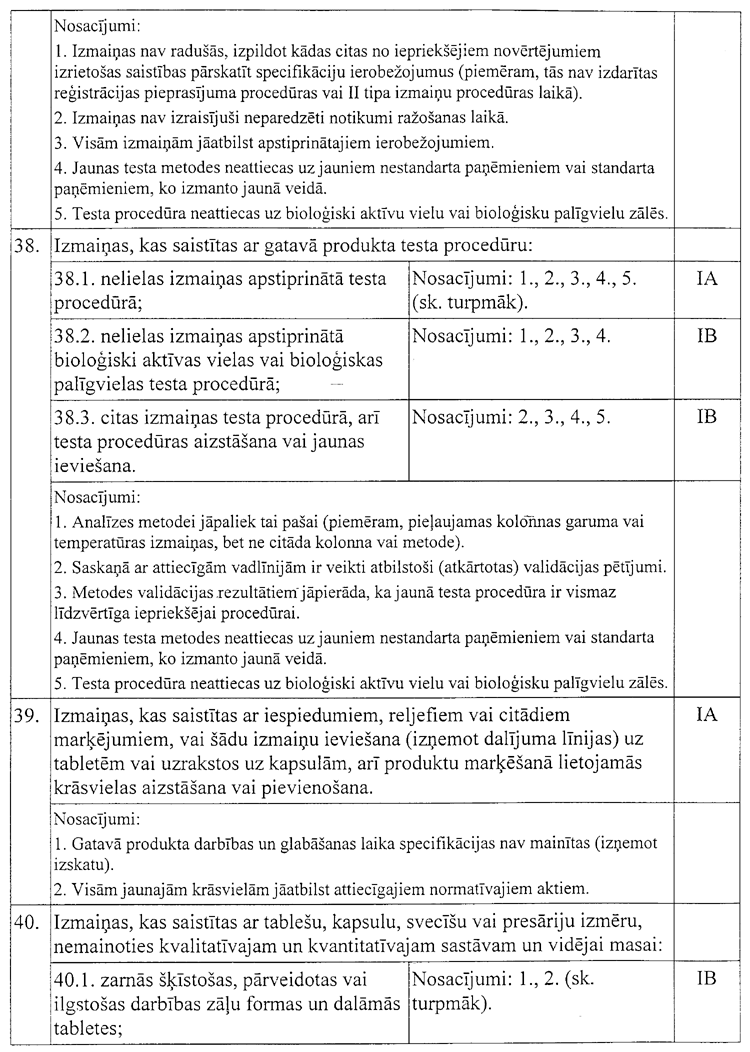
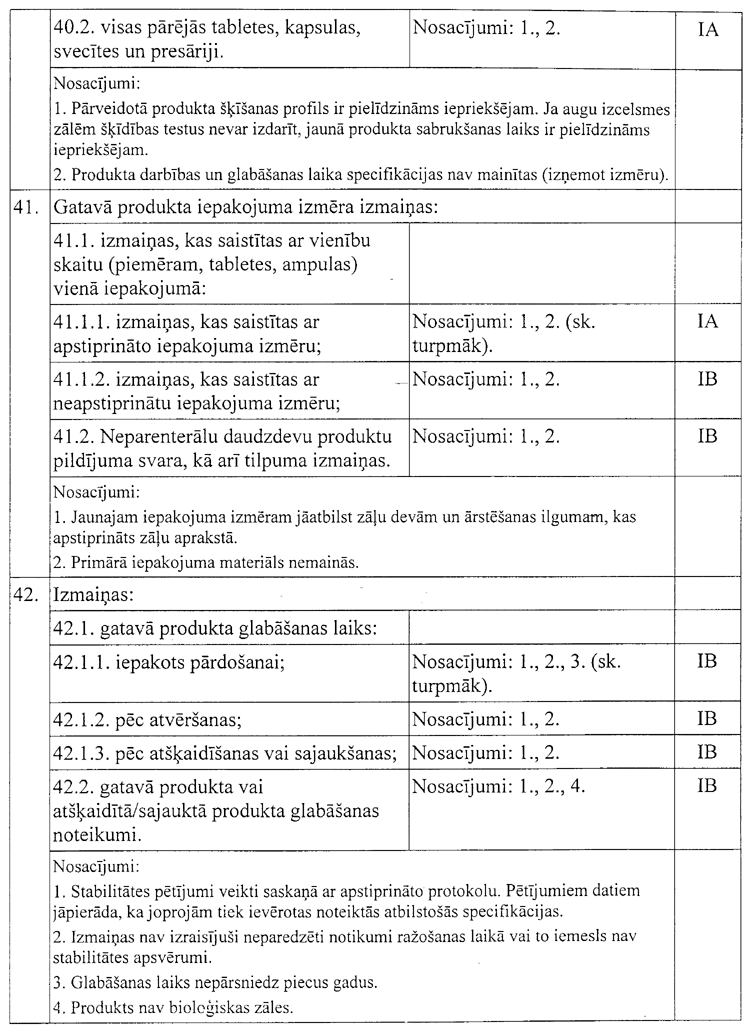
 "
"
"5.pielikums
Ministru kabineta
2000.gada 31.oktobra
noteikumiem Nr.381
 ".
".
Ministru prezidents
I.Emsis
Veselības ministrs
R.Muciņš
